
Abstract
Phosphoinositide-3-kinase, catalytic, alpha polypeptide, which encodes the catalytic p110α subunit of phosphatidylinositol 3-kinase α, is the most frequently mutated oncogene in human cancers. Targeting mutant p110α holds great promise for cancer therapy. However, it is challenging to develop p110α isoform-specific inhibitors. Most p110α mutations occur at two hot spot regions: an acidic cluster (E542, E545, and Q546) in the helical domain and a histidine residue (H1047) in the kinase domain. We recently discovered that p110α helical domain mutant proteins, but not the kinase domain mutant proteins, directly associate with insulin receptor substrate 1 (IRS1). Moreover, we demonstrated that disruption of protein–protein interaction between p110α helical domain mutant and IRS1 inhibits the growth of tumors with such mutations. The direct protein interaction between IRS1 and p110α helical domain mutants may provide a more accessible target for developing novel precision cancer therapy.
Keywords
Introduction
Targeted cancer therapy is transforming the way that doctors treat cancer patients. In contrast to standard chemotherapies that indiscriminately kill rapidly dividing cells in the human body, targeted cancer therapies are designed to affect specific molecules that are part of the pathways and processes used by cancer cells to grow, divide, and metastasize (Gschwind, Fischer, and Ullrich 2004). A major advantage of targeted therapies is that they elicit fewer toxic side effects than standard chemotherapies because they often cause little or no damage to normal cells. The best targets for cancer therapies are these molecules or pathways that are only present in cancer cells but not in normal cells (Gschwind, Fischer, and Ullrich 2004). For example, genetically altered proteins resulting from somatic mutations and translocations are ideal targets. A paradigm of targeted cancer therapy is the successful treatment of chronic myelogenous leukemia (CML) with Imatinib (branded as Gleevec; Druker et al. 2001). Over 90% of CML patients harbor a reciprocal translocation between chromosome 9 and 22, which results in an in-frame gene fusion between breakpoint cluster region (BCR) and Abelson murine leukemia virus (ABL1) gene. Imatinib inhibits the tyrosine kinase activity of BCR-ABL fusion protein, thereby killing only CML cells but not normal cells (Druker et al. 2001). Recently, the Food and Drug Administration approved Vemurafenib, a small molecule that inhibits mutant B-raf kinase activity, for treating metastatic melanoma patients with a BRAF V600E mutation. This success highlights the importance of targeting mutant oncoproteins for cancer therapy (Sosman et al. 2012). Given that recent cancer genome sequencing studies reveal that phosphoinositide-3-kinase, catalytic, alpha polypeptide (PIK3CA) is the most frequently mutated oncogene in human cancers (Garraway and Lander 2013), the mutant PIK3CA gene product, p110α, is an important target for cancer therapy. Here, we review the promises and challenges of targeting mutant p110α.
The PI3K/AKT Pathway Is a Central Intracellular Signaling Pathway
Phosphatidylinositol 3-kinases (PI3Ks) play key roles in regulating cell proliferation, survival, and motility (Figure 1). The PI3K family proteins are classified into three groups (see more detailed discussion below). The class I PI3Ks consist of a catalytic p110 subunit and a regulatory p85 subunits. In the basal state, the regulatory p85 subunit stabilizes the catalytic p110 subunit and inhibits its enzymatic activity (Liu et al. 2009). Upon growth factor stimulation, the Src Homology 2 (SH2) domains of p85 bind to the phosphotyrosine residues on the receptor protein kinases or adaptor proteins such as insulin receptor substrate 1 (IRS1), thereby activating the lipid kinase activity of PI3Ks (Cantley 2002). Activated PI3Ks convert phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 then recruits proteins that contain a pleckstrin homology domain (PH domain) including AKT serine/threonine kinase and 3-phosphoinositide-dependent kinase-1 (PDK1) to the cell membrane (Cantley 2002). The active membrane associated AKT and PDK1 phosphorylate numerous protein targets including mouse double minute 2 homolog (MDM2), glycogen synthase kinase 3 beta, IkappaB kinase, tubrin, and a subset of forkhead transcription factors (Cantley 2002). The tumor suppressor gene phosphatase and tensin homolog deleted on chromosome ten (PTEN) catalyzes the dephosphorylation of PIP3 to generate PIP2, thereby negatively regulating the PI3K-signaling pathways (Li et al. 1997). Inactivating mutations of PTEN have been found to occur in several different tumor types (Li et al. 1997).
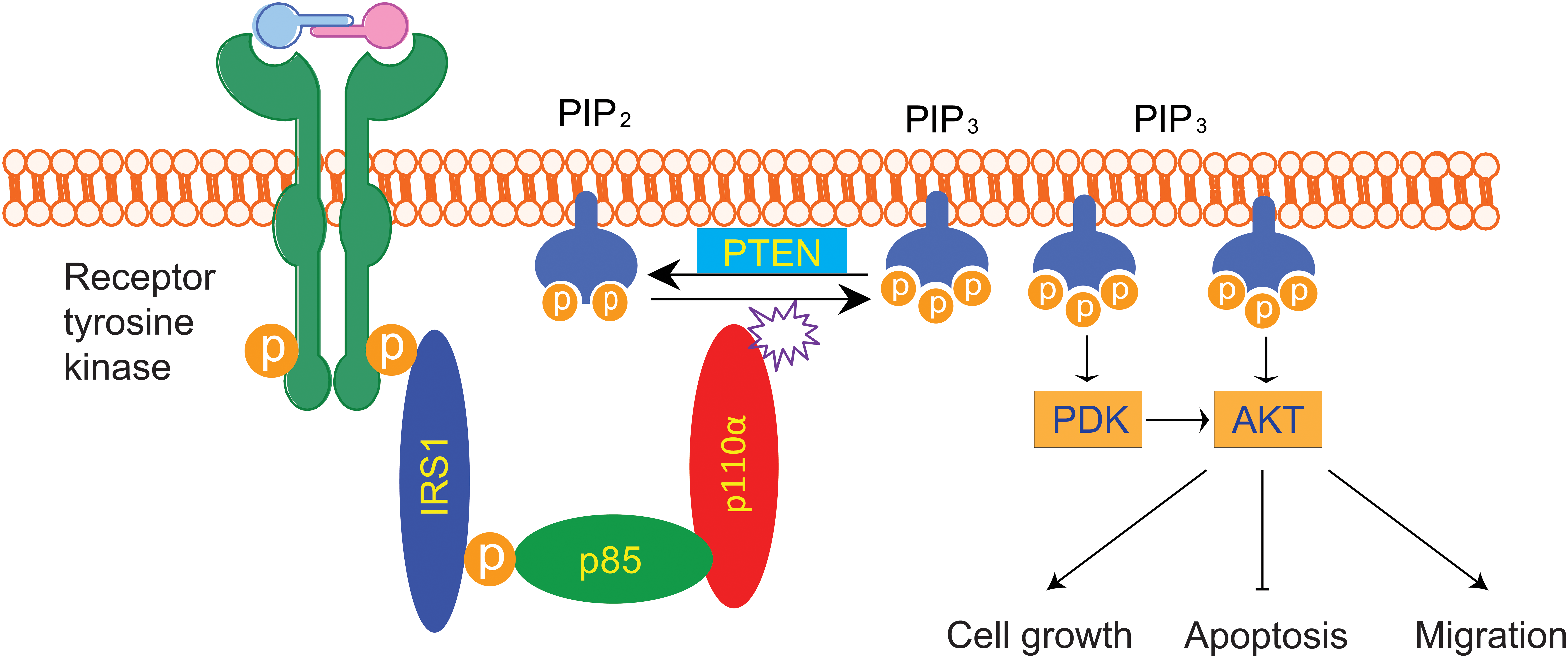
The PI3K/AKT-signaling pathway. Growth factor stimulation leads to activation of receptor protein tyrosine kinases, which in turn phosphorylate adaptor proteins such as IRS1. Phospho-IRS1 then recruits the PI3K heterodimers (p85 regulatory and p110 catalytic subunits) and activates the lipid kinase activity. The activated PI3Ks convert PIP2 to PIP3. The second messenger PIP3 recruits PDK and AKT to cytoplasmic membrane, where PDK phosphorylates and activates AKT. Activated AKT phosphorylates an array of downstream targets that control cell proliferation, apoptosis, and cell migration. IRS1 = insulin receptor substrate 1; PDK, 3-phosphoinositide-dependent kinase; PI3K, phosphatidylinositol 3-kinase; PIP2 = phosphatidylinositol-4,5-bisphosphate; PIP3 = phosphatidylinositol-3,4,5-triphosphate.
The Discovery of PIK3CA/p110α Mutation in Human Cancers Provides an Excellent Target for Cancer Therapy
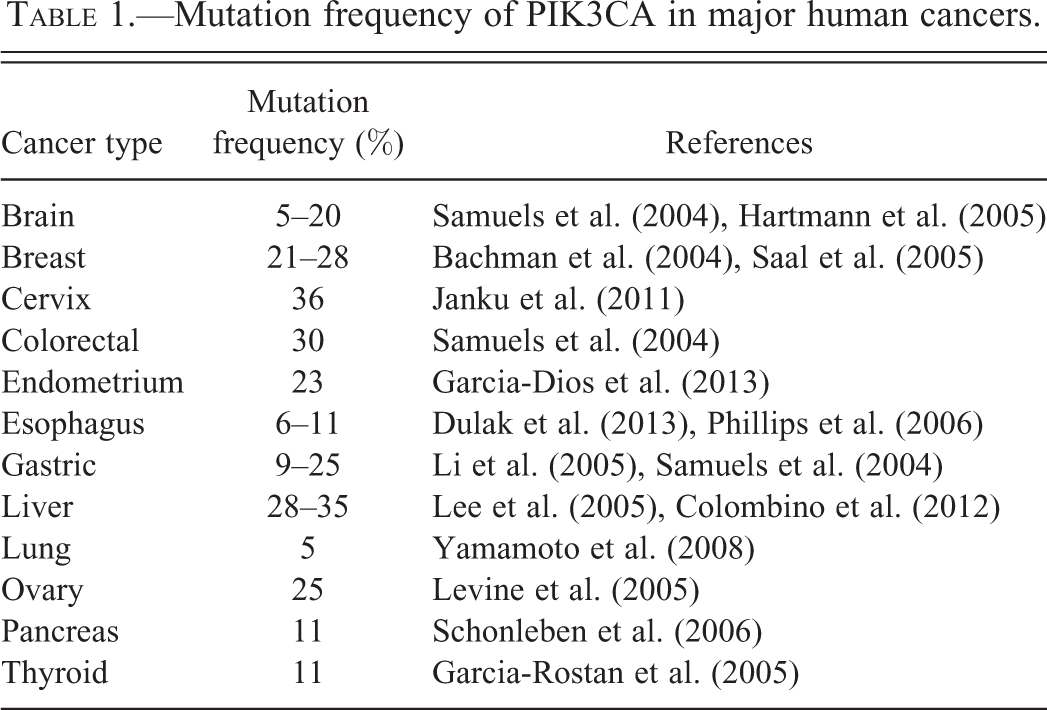
In the mutational analysis of PI3K gene family, Samuel et al. discovered almost a decade ago that PIK3CA (encoding p110α) was mutated in a variety of human cancers including brain, breast, colon, gastric, and lung cancers (Samuels et al. 2004). Follow-up studies by many laboratories and recent comprehensive cancer genome sequencing of a large collection of various human cancers by The Cancer Genome Atlas project revealed that PIK3CA is the most frequently mutated oncogene in human cancers (Table 1; Bachman et al. 2004; Broderick et al. 2004; Campbell et al. 2004; Lee et al. 2005; Levine et al. 2005; Li et al. 2005; Oda et al. 2005; Garraway and Lander 2013). Interestingly, nearly all of the mutations are heterozygous and the majority are clustered in at two hot spot regions: an acidic cluster (E542, E545, and Q546) in the helical domain and a histidine residue (H1047) in the kinase domain (Figure 2). Results from in vitro lipid kinase assays indicated that the hot spot mutations, H1047R and E545K, resulted in increased lipid kinase activity, suggesting that the mutant p110α indeed acts as an oncogene (Samuels et al. 2004). Targeted inactivation of the PIK3CA oncogenic mutations in human colorectal cancer (CRC) cell lines showed that oncogenic alleles of PIK3CA render cancer cells growth advantage in low serum condition, attenuation of apoptosis, and facilitating tumor invasion (Samuels et al. 2005). Interestingly, mutant p110α appears to selectively activate AKT1 and the forkhead transcription factors FKHR and FKHRL1 (Samuels et al. 2005). Given that the oncogenic mutant p110α is a lipid kinase, it is a very promising drug target for cancer therapy as it is actionable, but a potential limitation is specificity.
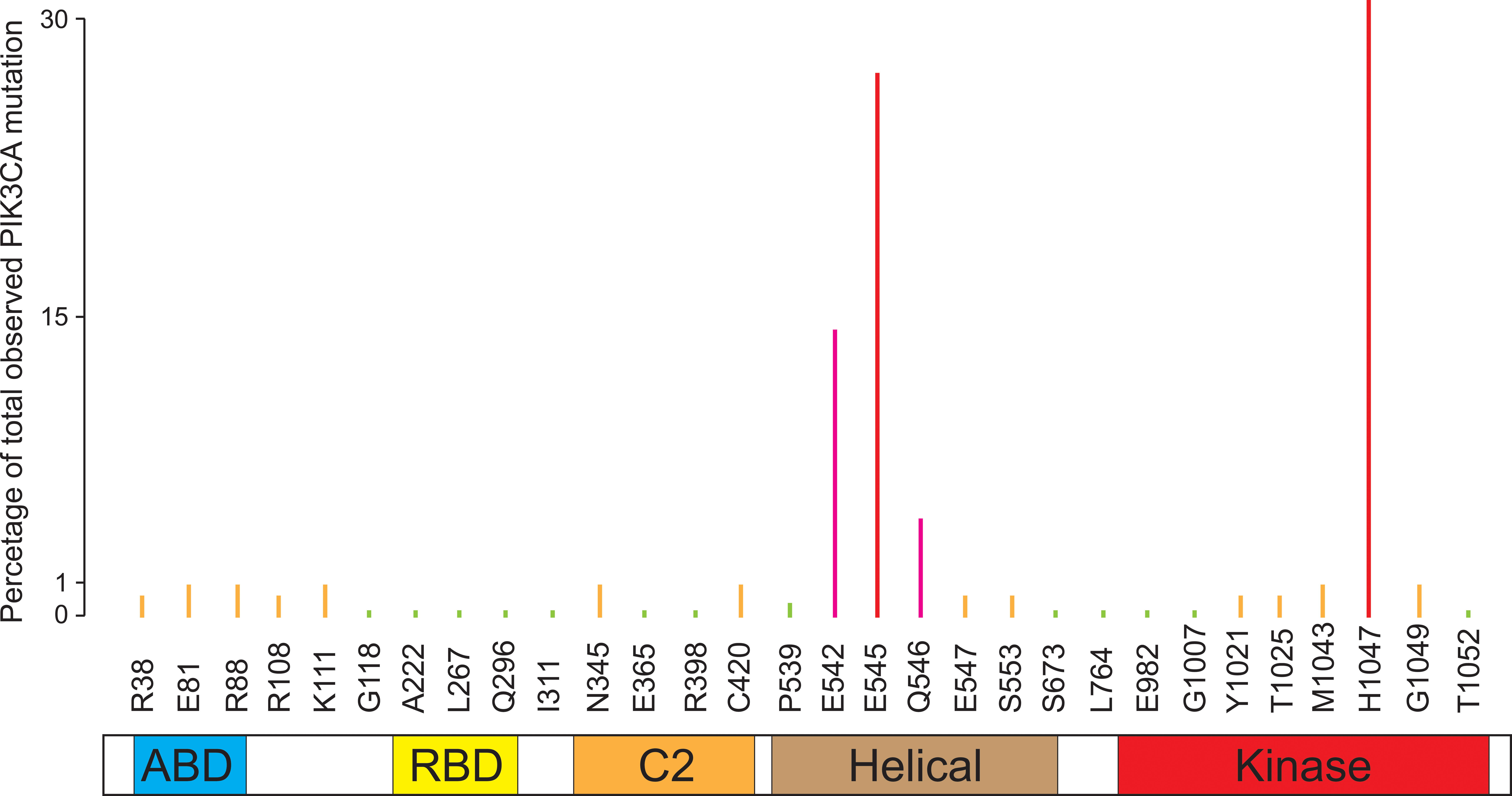
Somatic mutation of PIK3CA/p110α in human cancers. The p110α subunit consists of several different domains: ABD: adapter binding domain (AKA: p85 binding domain); C2: C2 domain; helical: helical domain; kinase: kinase domain; PIK3CA = phosphoinositide-3-kinase, catalytic, alpha polypeptide; RBD: Ras-binding domain.
Mutation frequency of PIK3CA in major human cancers.
Challenges of Targeting Mutant p110α
Although scientists at both pharmaceutical companies and academic institutions have made enormous effort to develop drugs to inhibit mutant p110α enzymatic activity, it turns out that it is difficult to find small molecules that specifically inhibit p110 isoforms (Liu et al. 2009). Based on the substrate specificities and structural features, PI3K family members are divided into three classes (Figure 3). Class I PI3Ks are heterodimers consisting of a p110 catalytic subunit and a p85 regulatory subunit. The p85 regulatory subunit modulates the activity, stability, and localization of the enzyme. Three genes, PIK3R1, PIK3R2, and PIK3R3, encode p85α (and its splice variants p55α and p50α), p85β, and p55γ, respectively. All these regulatory subunits are collectively referred to as p85. The class I PI3Ks convert PIP2 to PIP3. Both class II and class III PI3K family members have only a single catalytic subunit. The class II PI3Ks (PIKC2α, PIKC2β, and PIKC2γ) use phosphatidylinositol or phosphatidylinositol-4-phosphate as substrates; whereas the single class III PI3K member, VPS34, converts phosphatidylinositol to phosphatidylinositol-3-phosphate.
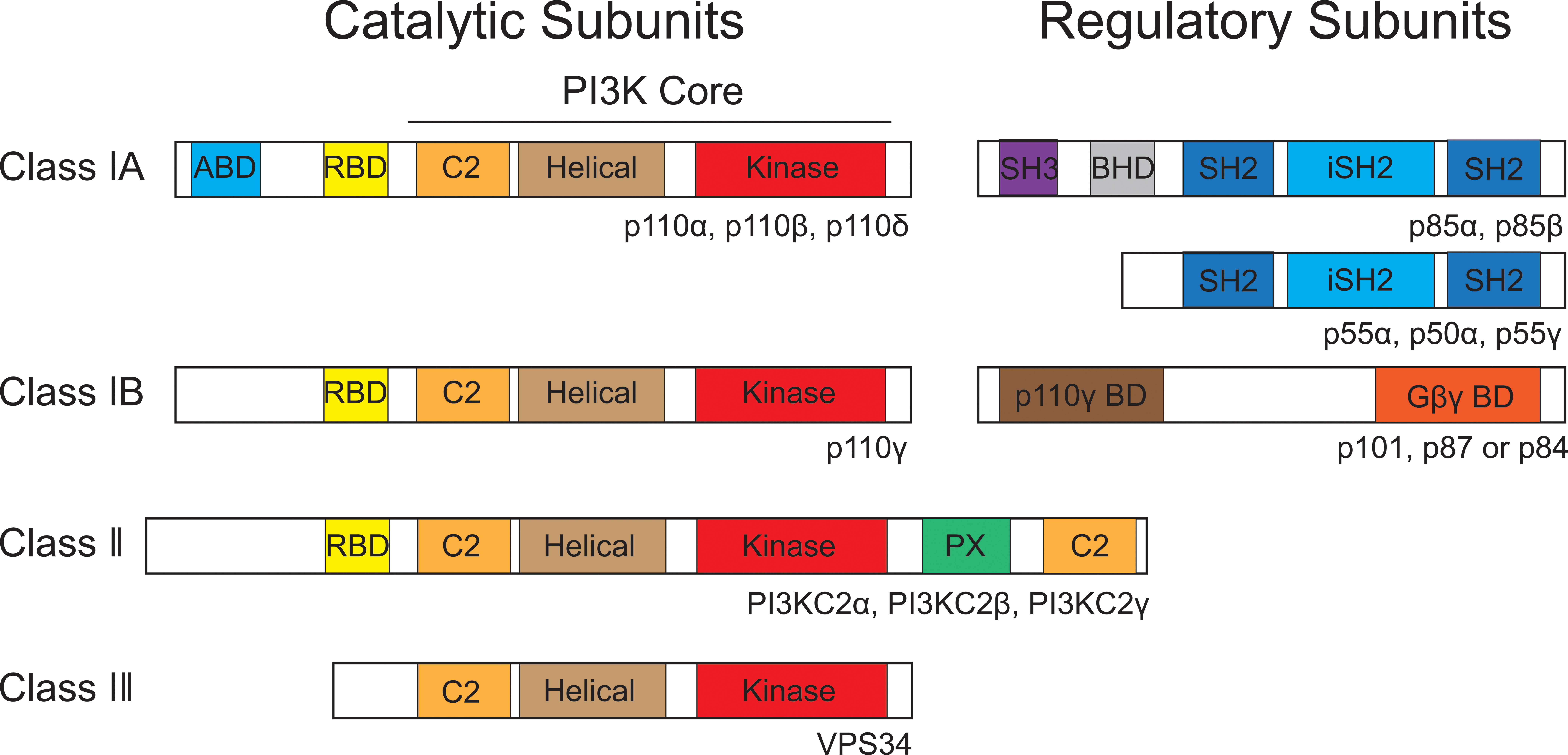
Structures of PI3Ks. PI3Ks are classified into three groups: class I, class II, and class III. Class I PI3Ks are heterodimers consisting of a catalytic p110 subunit and a p85 regulatory subunit. The class I PI3Ks are further divided into class IA and class IB subgroups. Class IA PI3Ks consist of PI3Kα, PI3Kβ, and PI3Kδ with p110α, p110β, and p110δ as their catalytic subunits, respectively. The catalytic subunit can form a heterodimer with any of the p85 subunit (p85α, p85β, p55α, p50α, and p55γ). The class IB PI3K subgroup consists of a p110γ catalytic subunit and one of the regulatory subunits (p101, p87, and p84). The class II and III PI3Ks consist of only a catalytic polypeptide. Several different domains are present including BHD: BCR homology domain; Gβγ BD: G beta gamma protein complex binding domain; iSH2: inter-Src homology 2 domain; p110γ BD: p110γ binding domain; PI3Ks, phosphatidylinositol 3-kinase; PX: phosphoinositide-binding structural domain; SH3: Src homology 3 domain; SH2: Src homology 2 domain.
The class I PI3Ks have been intensively characterized. This group of PI3Ks is further divided into class IA and class IB subgroups. The class IA PI3Ks have three members: PI3Kα, PI3Kβ, and PI3Kδ. The catalytic subunits of these three enzymes share similar domain structures: an N-terminal adaptor-binding domain (ABD, also called p85 binding domain), a Ras-binding domain (RBD), a C2 domain, a helical domain, and a catalytic domain (Figure 3). The p110 catalytic subunit of the class IA PI3Ks forms a heterodimer with one of the p85 regulatory subunits (p85α, p85β, p55α, p55γ, and p50α). The class IB PI3K consists of the catalytic subunit p110γ and regulatory subunit p101 (Figure 3). The overall domain structure of p110γ is similar to those of PI3Kα, PI3Kβ, and PI3Kδ, but it lacks the ABD domain (Figure 3). While p110α and p110β are ubiquitously expressed, p110δ and p110γ are mainly expressed in leukocytes (Liu et al. 2009). Interestingly, only p110α is mutated in human cancers (Zhao and Vogt 2008a). Recent crystal structure analyses indicate that the structures of class I PI3K catalytic domains are quite similar and that the ATP-binding pockets of these enzymes are almost identical (Vadas et al. 2011), which may explain why it is difficult to develop p110α isoform-specific inhibitors.
Currently, several pan-class I PI3K inhibitors (GDC-0941, BKM120, PX866, and BAY 80-6946) and dual PI3K/mTOR inhibitors (e.g., BEZ235) are in early-stage clinical trials (Rodon et al. 2013). There are also some PI3Kβ-specific and PI3Kδ-specific inhibitors under clinical evaluation for their efficacy against various tumor types (Rodon et al. 2013). It is worth noting that Genentech has recently developed a so-called β-spare PI3K inhibitor GDC-0032, which selectively inhibits p110α, p110δ, and p110γ but sparing the p110β isoform. Early-stage clinical trials in breast cancer patients have shown great promise. Patients harboring PIK3CA/p110α had at least partial clinical responses to the β-spare PI3K inhibitor (D. Juric, personal communication, April 9, 2013), suggesting that a p110α isoform-specific inhibitor should be more potent and less toxic. Nonetheless, drugs that specifically target the mutant p110α should be highly advantageous.
Mutant-specific Protein Interaction between IRS1 and Helical Domain Mutations of p110α May Provide a More Accessible Target for Cancer Therapy
Given the challenges encountered with the development of p110α-specific inhibitors, alternative approaches are needed to target mutant p110α for cancer therapy. Our recent discovery that the helical domain p110α mutants directly interact with IRS1 may provide a more accessible target for developing drugs to treat cancer patients harboring those p110α mutations (Hao et al. 2013). We discuss below the relevant background and therapeutic implications of our findings.
The p110α Helical Domain and Kinase Domain Mutations Exert Their Oncogenic Functions through Distinct Mechanisms
As mentioned above, most of the p110α mutations occur at two hot spot regions: an acidic cluster (E542, E545, and Q546) in the helical domain and a histidine residue (H1047) in the kinase domain. The E545K and H1047R are the two most frequently observed p110α somatic mutations in human cancers. Interestingly, several recent studies indicate that the E545K and H1047R mutations exert their oncogenic functions through distinct mechanisms: (1) Zhao and Vogt (2008b) have shown that the helical domain and the kinase domain mutant proteins could synergistically transform chicken embryonic fibroblasts, providing the first clue that the two mutations may induce oncogenic transformation through different pathways. Consistently, they further demonstrated that the p110α helical domain mutants require the RBD for their transformation activities, whereas the oncogenic activity of the H1047R mutant depends on the ABD domain. (2) Pang et al. (2009) showed that expression of the p110α E545K mutant produce a more severe metastatic phenotype than that of the H1047R mutant in a breast cancer cell line. (3) In contrast, the p110α H1047R mutant, but not the E545K mutant, is found to enhance human epidermal growth factor receptor 2–mediated transformation of immortalized mammary epithelial cells (Chakrabarty et al. 2010). Structural analysis indicates that the p110α H1047R mutation alters the interaction between PI3Kα and the cell membrane, thereby activating its kinase activity (Mandelker et al. 2009). It has been suggested that the helical domain mutations activate their enzymatic activities by disrupting the inhibitory effect of the p85 subunits (Huang et al. 2007; Miled et al. 2007).
Direct Interaction between p110α Helical Domain Mutants and IRS1 Drives Tumorigenesis
Our recent studies, however, indicate that the weakened p110α-p85 interaction caused by the p110α helical domain mutations is not sufficient for these mutant proteins to exert their oncogenic functions. In an attempt to determine how p110α E545K and p110α H1047R differentially activate oncogenic-signaling pathways, we tried to identify proteins that might differentially bind to wild-type (WT) and mutant p110α. To this end, we discovered that IRS1 binds to p110α E545K mutant but not to WT or H1047R mutant under serum-starvation conditions (Hao et al. 2013). In addition, other hot spot p110α mutations in the helical domain including E542K, E545A, E545G, and Q546K as well as relatively rare mutations including K111N in the ABD domain and N345K in the C2 domain gain interaction with IRS1 under serum-starvation conditions (Hao et al. 2013). We provided compelling evidence that the p110α E545K mutant-IRS1 interaction plays a critical role in tumorigenesis and demonstrated how this mutant-specific protein interaction may be exploited for cancer therapy. First, in the IRS1 knockout (KO) DLD1 cells harboring the p110α E545K mutation, reconstitution of an IRS1 deletion construct that does not interact with p110α E545K mutant protein produces smaller xenograft tumors compared to the full-length IRS1 reconstituted cells (Hao et al. 2013). Second, a p110α E545K mutant stapled peptide, but not the WT counterpart, specifically inhibits xenograft tumor growth of CRC cells with a p110α E545K mutation (Hao et al. 2013). Mechanistically, our data suggest that p110α E545K mutant-IRS1 interaction activates the mutant p110α lipid kinase in two ways: (1) It brings the mutant enzyme complexes from cytosol to plasma membrane; (2) it stabilizes the mutant p110α proteins (Hao et al. 2013). We propose that the helical domain mutations of p110α, as well as some of the mutations in the ABD and C2 domain, induce conformational changes that enable p110α to directly interact with IRS1, which does not require IRS1 tyrosine phosphorylation and p85 proteins (Figure 4). In support, a recent study shows that oncogenic mutations of p110α in various domains indeed induce conformational changes in its protein structure that may affect its structural interactions (Burke et al. 2012).
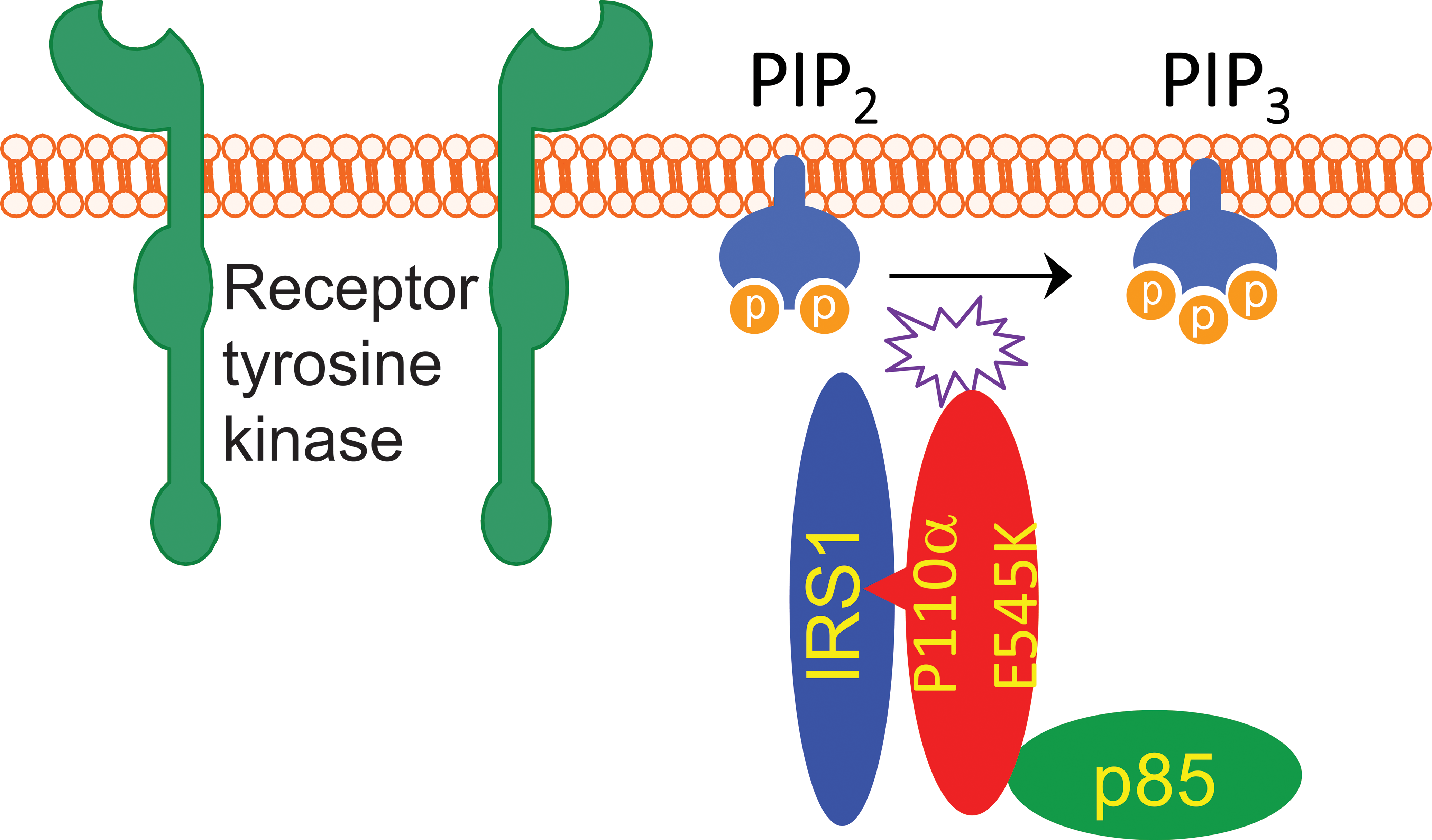
Protein interaction between helical domain mutation of p110α and IRS1 drives tumorigenesis. In cancer cells harboring p110α helical domain mutations, IRS1 directly binds to mutant p110α and brings it the cell membrane, thereby activating the PI3K-AKT-signaling pathway independent of any stimulation by growth factor. IRS1, insulin receptor substrate 1.
Our studies may provide an explanation for observed different phenotypes produced by the helical and kinase domain mutations. Using isogenic breast cancer cell lines, Pang and colleagues (2009) demonstrated that the expression of p110α helical domain mutations increase the sensitivity of cancer cells to chemoattractants and induce much stronger metastatic phenotypes than the H1047R kinase domain mutation. These phenotypic differences cannot be simply explained by the increased enzymatic activities of the helical and kinase domain mutations. In fact, Vasudevan et al. (2009) observed that the basal levels of phospho-AKT are lower in cancer cell lines expressing p110α helical domain mutations compared to these cell lines expressing the H1047R kinase domain mutation. Consistently, when complexed with p85 regulatory subunit, the p110α H1047R kinase domain mutant protein displays higher lipid kinase activity in vitro than the E545K helical domain mutant (Samuels et al. 2005). We showed that the p110α helical domain mutants, but not the kinase domain mutant, directly associate with IRS1 without growth factor stimulation (Hao et al. 2013). In this regard, we suggest that the p110α helical domain mutant-IRS1 protein complexes may contain additional proteins that do not exist in the p110α kinase domain mutant protein complexes, thereby producing distinct phenotypes.
The Protein Interaction between IRS1 and p110α Helical Domain Mutants Is Independent of the p85 Regulatory Subunit
Given that p110α is brought to the IRS1 complex through interaction between phospho-IRS1 and p85 regulatory subunit when cells are stimulated with growth factor, it is important to demonstrate that the p110α helical domain mutant-IRS1 interaction is independent of p85. Our studies provided several lines of evidence indicating that interaction between the p110α helical domain mutant proteins and IRS1 is not mediated by binding of p85 SH2 domain with pY residues on IRS1. First, p110α helical domain mutant-IRS1 interaction is independent of IRS1 tyrosine phosphorylation (Hao et al. 2013). Second, a p110α E545K mutant protein (p110α E545KΔABD) devoid of the ability to bind with p85 proteins still interacts with IRS1 (Hao et al. 2013). This result also provides an explanation for the observation made by Vogt and colleagues that the helical domain mutants of p110α do not require the binding of p85 to transform chicken embryonic fibroblasts (Zhao and Vogt 2008b). Third, we show here that ablation of p85 proteins does not reduce the p110α E545K mutant-IRS1 interaction (Hao et al. 2013). Interestingly, diminishment of p85 proteins actually enhances the p110α E545K mutant-IRS1 interaction, suggesting that p85 may compete with IRS1 to bind to the p110α helical mutants (Hao et al. 2013). Fourth, in the absence of p85 proteins, recombinant IRS1 proteins bind in vitro to p110α E545K mutant but not WT p110α proteins (Hao et al. 2013). Fifth, an IRS1 deletion that abrogates IRS1 interaction with p110α E545K mutant still binds well with p85 proteins (Hao et al. 2013). Sixth, a p110α E545K mutant peptide, which disrupts the p110α E545K mutant-IRS1 interaction, does not affect the binding of the mutant p110α to p85 (Hao et al. 2013). However, we point out that even though p85 proteins are not required for direct interaction between the p110α helical domain mutants and IRS1, they may participate in an interaction that provides stability to the mutant because ablation of p85 proteins results in significant reduction of the levels of p110α protein (Hao et al. 2013).
Disrupting Protein–Protein Interaction between p110α Helical Domain Mutant and IRS1 Provides a Novel Approach for Development of Drugs Targeting Cancers with These Mutations
Increasing evidence suggests that protein–protein interactions are druggable targets (White, Westwell, and Brahemi 2008). In fact, small molecules that inhibit the MDM2-p53 interaction are being tested in clinical trials to treat cancer patients (Essmann and Schulze-Osthoff 2011). Moreover, ABT-737, a small molecule B-cell lymphoma 2 (BCL2) homology domain 3-mimetic that inhibits protein–protein interaction of BCL2 family proteins, is also being evaluated in clinical trials for its efficacy in hematologic malignancies (Khaw, Huang, and Roberts 2011). Our observation that a p110α E545K mutant staple peptide, but not the WT counterpart, specifically inhibits xenograft tumor growth of CRC cells with a p110α E545K mutation provides a proof of principle for targeting this mutant-specific protein interaction for cancer therapy. The discovery of frequent mutations of PIK3CA in human cancer provides a strong rationale for inhibition of mutated p110α activities for targeted cancer therapy (Samuels et al. 2005). However, as discussed above, it has remained a challenge to develop p110α isoform-specific inhibitors. Our data suggest that disruption of the interactions between helical domain p110α mutants and IRS1 may be exploited as a more accessible targeted therapy approach for cancer patients harboring such mutations. Given that this mutant p110α-specific interaction exists only in tumor cells that harbor these particular mutations, but not in normal tissues, drugs targeting this cancer-specific interaction should have no or minimal side effects because they do not perturb normal cellular functions. It is worth noting that our p110α E545K mutant stapled peptide is not stable in the blood stream, which prevents it being delivered systematically. A mean to improve the pharmacokinetics of the peptide is to substitute the peptide bonds with chemical bonds to generate a functional peptidomimetics. Alternatively, small molecules that can disrupt protein–protein interaction between IRS1 and p110α helical domain mutant could be identified through high-throughput screening.
Prospective
Mutant p110α is an excellent target for cancer therapy. Much of current effort has been devoted to develop p110α-isoform-specific inhibitors, which turns out to be a challenge task. Our recent finding that p110α helical domain mutants gain direct interaction with IRS1 raises new hope of targeting this mutant-specific protein interaction for cancer therapy. The key will be to screen or design small molecules with favorable pharmacokinetics that disrupt the protein–protein interaction between p110α helical domain mutant and IRS1. In this regard, a co-crystal structure of p110α helical domain mutant and IRS1 would be very helpful for both in silico design and optimization of compounds that disrupts the p110α mutant-specific protein–protein interaction. Finally, drugs targeting mutant p110α may also be used to treat hemimegalencephaly, because recent next generation sequencing analyses revealed that p110α is also mutated in patients with this disease (Lee et al. 2012).
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by NIH grants R21CA160060, R01CA127590, R01HG004722, P50CA150964, and P30 CA043703.