
Abstract
Compound-induced pancreatic injury is a serious liability in preclinical toxicity studies. However, its relevance to humans should be cautiously evaluated because of interspecies variations. To highlight such variations, we evaluated the species- and dose-specific pancreatic responses and progression caused by GI181771X, a novel cholecystokinin 1 receptor agonist investigated by GlaxoSmithKline for the treatment of obesity. Acute (up to 2,000 mg/kg GI181771X, as single dose) and repeat-dose studies in mice and/or rats (0.25–250 mg/kg/day for 7 days to 26 weeks) showed wide-ranging morphological changes in the pancreas that were dose and duration dependent, including necrotizing pancreatitis, acinar cell hypertrophy/atrophy, zymogen degranulation, focal acinar cell hyperplasia, and interstitial inflammation. In contrast to rodents, pancreatic changes were not observed in cynomolgus monkeys given GI181771X (1–500 mg/kg/day with higher systemic exposure than rats) for up to 52 weeks. Similarly, no GI181771X treatment-associated abnormalities in pancreatic structure were noted in a 24-week clinical trial with obese patients (body mass index >30 or >27 kg/m2) as assessed by abdominal ultrasound or by magnetic resonance imaging. Mechanisms for interspecies variations in the pancreatic response to CCK among rodents, monkeys, and humans and their relevance to human risk are discussed.
Keywords
Introduction
Cholecystokinin (CCK), a peptide hormone of the gastrointestinal system and brain, has numerous and critical physiological roles, including pancreatic enzyme secretion, stimulation of gallbladder contraction, slowing of gastric emptying, induction of satiety, and suppression of food intake (Reviewed in Cawston and Miller 2010; Chandra and Liddle 2011; Rehfeld 2011; Duca and Covasa 2012). CCK is synthesized and secreted by enteroendocrine I cells of the small intestine in response to nutrient stimulation, specifically by fat and protein in the chyme. CCK-mediated satiety has been well documented in both animals and humans after the discovery of the two CCK receptors, that is, CCK1 (cholecystokinin 1 receptor [CCK1R]) and CCK2 (CCK2R) receptors (originally designated as CCKA and CCKB receptors, respectively). CCK1 receptors are expressed in pancreas, gallbladder, stomach, vagus nerve, small intestine, and hypothalamus, including paraventricular and dorsomedial nuclei (Berna and Jensen 2007; Wang and Cui 2007). CCK mediates its satiety through the CCK1R by a paracrine mode of action on vagal afferent neurons to the central nervous system (Ritter, Covasa, and Matson 1999). CCK-induced satiation can be attenuated in rats by vagotomy or by deactivation of vagal afferents with capsaicin, indicating that the CCK1R relays the postprandial satiety signal via the vagal afferent neurons to the central nervous system (Ritter and Ladenheim 1985; Smith, Jerome, and Norgren 1985). CCK1-receptor-activated satiation has been documented in rat studies, whereas CCK2 receptors are likely not involved in the satiation of CCK as evidenced by data obtained from the Otsuka Long-Evans Tokushima Fatty (OLETF) rats that are lacking active CCK1 receptors (Takiguchi et al. 1997; Moran et al. 1998). The finding that exogenous CCK reduces meal size in rats has been replicated in many laboratories and extended to numerous species, including nonhuman primates and humans (Gibbs, Young, and Smith 1973; Gibbs, Falasco, and McHugh 1976; Kissileff et al. 1981; Pi-Sunyer et al. 1982; Stacher et al. 1982; Muurahainen et al. 1991; Lieverse et al. 1995; Greenough at al. 1998; Macintosh et al. 2001). Consistent with this satiety effect, CCK1 antagonists increase meal size in experimental animals and humans (Reidelberger and ORourke 1989; Reidelberger, Varga, and Solomon 1991; Moran et al. 1992, 1993). To take therapeutic advantage of CCK1R’s role in satiety, GI181771X, a novel CCK1R agonist was investigated by GlaxoSmithKline for the treatment of obesity. GI181771X is a 1,5-benzodiazepine that reduces food intake in rats presumably by activation of peripheral CCK1R on vagal afferent neurons and on the pyloric sphincter (Moran et al. 1990; Cox and Randich 1997). GI181771X is devoid of classic benzodiazepine properties and is also an antagonist for CCK2R. However, unlike octapeptide (CCK-8), which requires parenteral administration, GI181771X demonstrates efficacy after oral administration.
In addition to CCK’s role in satiation and weight control, CCK1R agonism has the potential to cause pancreatic disorders due to CCK1R hyperstimulation and abnormal exocrine secretion. CCK directly stimulates CCK1R on acinar cells and induces pancreatic enzyme secretion. CCK also binds CCK1R on vagal and intrapancreatic neurons modulating pancreatic exocrine secretion via a cholinergic mechanism (i.e., release of acetylcholine; reviewed in Weiss, Halangk, and Lerch 2008). Recently, pancreatic stellate cells from rats and humans have been shown to express CCK1R and CCK2R and synthesize and release acetylcholine, which in turn causes exocrine secretion via stimulation of muscarinic receptors (M1 and M3 receptors) (Berna et al. 2010; Phillips et al. 2010). CCK therefore stimulates pancreatic exocrine secretion both directly via CCK1R and indirectly via muscarinic receptors. An area of interest is the role of exaggerated CCK1R hyperstimulation in the pathogenesis of pancreatitis. Several lines of evidence indicate that CCK or CCK1R agonists cause pancreatitis in animals. Cerulein, a CCK analog, causes pancreatitis in many animals including mice, rats, rabbits, dogs, and pigs (Willemer, Elsasser, and Adler 1992; Gorelick, Adler, and Kerin 1993). Pancreatitis induced by high doses of cerulein in rats and mice is a widely used experimental model, and the histological presentation of this model is quite similar to the early phase of acute pancreatitis in humans (Longnecker and Wilson 2002; Green and Reeve 2008). Prolonged and sustained CCK-induced secretions may result in misdirection and autodigestion of adjacent tissues, disrupting the integrity of acini and their attendant ducts (Gaisano and Gorelick 2009). It has been shown that the disturbance of acinar–ductal relationship plays an important role in the establishment of chronic pancreatitis, and increased ductal proliferation/replication is a consistent feature of chronic pancreatitis (Taguchi, Yamaguchi, and Otsuki 2002; Stevens, Conwell, and Zuccaro 2004; Whitcomb 2004; Bhanot and Moller 2009) in humans. Additionally, local tissue damage and the subsequent inflammatory stimulation may induce recruitment of somatic stem cells to permit repair, and increased ductal replication has also been proposed as a contributing factor of chronic pancreatitis by distorting and obstructing small pancreatic ducts (Stevens, Conwell, and Zuccaro 2004; Whitcomb 2004; Bhanot and Moller 2009). Similarly, repetitive injury induced by cerulein treatment has been shown to cause chronic pancreatitis in mice that resembled morphological characteristics of human chronic pancreatitis (Neuschwander-Tetri et al. 2000). In humans, low-grade chronic pancreatitis is a well-established risk factor for pancreatic adenocarcinoma (Jura, Archer, and Bar-Sagi 2005; Landi 2009), a serious clinical outcome with a 5-year life expectancy. Increased ductal replication due to chronic pancreatitis is considered to be the first stage of progression to pancreatic adenocarcinoma (Jura, Archer, and Bar-Sagi 2005). While risk of chronic pancreatitis progressing to pancreatic cancer increases with duration of pancreatitis, the majority of patients with chronic pancreatitis do not develop pancreatic cancer.
In humans, pancreatitis is a life-threatening condition with significant morbidity and mortality and, therefore, establishing the relevance to humans of compound-induced pancreatic injury in preclinical toxicity studies is essential because of species variation in pancreatic pathophysiology. To highlight such variations, we evaluated the species- and dose-specific pancreatic responses caused by GI181771X, a CCK1R agonist investigated by GlaxoSmithKline for the treatment of obesity. We describe here the species- and dose-specific pancreatic responses to GI181771X administration in CD-1 mice, Han Wistar rats, and Cynomolgus monkeys.
Materials and Methods
All procedures involving animals conformed to the guidelines set forth in the Guide to the Use and Care of Laboratory Animals and were reviewed and approved by the GlaxoSmithKline Institutional Animal Care and Use Committee. The pancreatic findings were reviewed in single-dose studies in mice and rats, and repeat-dose studies in rats (7-day, 4-week, and 26-week) and monkeys (4-week, 26-week, and 52-week).
Acute Single-Dose Study in Mice and Rats and Repeat-Dose Studies in Rats
Rodent studies were performed to determine the maximum nonlethal dose/target organ toxicity following single oral dose administration of GI181771X (Table 1) or to assess toxicity associated with repeated oral administration of GI181771X for 7 days, 4 weeks, and 26 weeks (Table 2). Male and female animals were obtained from Taconic (Germantown, NY) or Charles River (Margate, UK), randomized to treatment groups using computer-generated tables of random numbers, housed by sex in polycarbonate solid-bottom cages, and maintained at 64°F to 79°F room temperature with 30% to 70% humidity and fluorescent 12-hr light–dark cycle. Animals were fed certified Rodent Diet (#5002; PMI Feeds Inc., St. Louis, MO) or Rat No. 1 Expanded Diet (Special Diets Services Ltd, UK), and water was provided ad libitum. In acute studies, animals were observed for 2 or 14 days after dose administration, and sacrificed on day 3 and day 15. Animals were sacrificed at the end of the dosing period in repeat-dose studies (Table 2). Blood samples for routine clinical pathology parameters were collected at necropsy (abdominal vena cava) and on various interim time points (caudal tail vein). Amylase and lipase activities were analyzed from serum (by enzymatic method in 7-day and 4-week studies) and plasma (by turbidimetric method in the 26-week study). Complete necropsies were performed and protocol tissues, including pancreata, were harvested or weighed and fixed in 10% neutral buffered formalin, processed, sectioned, and stained with hematoxylin and eosin (H&E) for histopathological evaluation.
Acute single-dose study in CD-1 mice and Han Wistar rats.

a Vehicle—Polyethylene glycol 400.
Repeat-dose studies in Han Wistar rats.

Note: GLP = Good Laboratory Practice.
a Vehicle—polyethylene glycol 400.
b Vehicle—0.5% (w/w) hydroxypropylmethylcellulose and 0.1% (w/w) Tween 80.
c Results of pancreatic findings.
Repeat-Dose Studies in Monkeys
Repeat-dose studies in cynomolgus monkeys (Macaca fascicularis) were conducted at Covance (Covance Laboratories Inc., 9200 Leesburg Pike, Vienna, VA), MPI (MPI Research, 54943 North Main Street, Mattawan, MI), and CRL (Charles River Laboratories, 87 Senneville Road, Senneville, QC, Canada; Table 3). Cynomolgus monkeys were received from Covance Research Products (Denver, PA). Following a minimum 30-day quarantine period, animals were acclimated to laboratory conditions for approximately 4 weeks prior to the initiation of dosing. During the acclimation period, the animals were tested for tuberculosis three times and given an anthelmintic (Ivomec®; Merck and Company, Whitehouse Station, NJ). Animals were individually housed in a stainless steel, rack-mounted cage and uniquely identified by a tattoo that was used during pretreatment evaluations and randomization. Certified primate diet (#8726C, Harlan Teklad, Madison, WI or Hi-Fiber Primate Diet 5K91; Purina Mills Nutrition International Inc., St. Louis, MO) was available twice daily except during designated procedures. Food was withheld overnight prior to the collection of samples for clinical pathology (including urinalysis) and prior to scheduled necropsy. During the treatment phase, animals were fed 1 hr prior to dosing. Tap water was available ad libitum. The temperature and relative humidity in the animal room were set to maintain the protocol range of 18°C to 29°C and 30% to 70%, respectively. During the study period, monkeys were monitored at least once daily. Ten or greater air changes/hour and a 12-hr light/12-hr dark cycle (lights on approximately 0600 to 1800 hr) were maintained. Blood samples for analysis of plasma GI181771 concentrations were obtained from all animals in each study group on various days per protocol. Animals were dosed with either the control article or appropriate test formulation via nasal gastric gavage once daily for 4 weeks, 26 weeks, or 52 weeks. Animals were sacrificed at the end of study. Complete necropsies were performed, and protocol tissues, including pancreata, were harvested or weighed and fixed in 10% NBF, processed, sectioned, and stained with H&E for histopathological evaluation.
Repeat-dose studies in cynomolgus monkeys.
Note: GLP = Good Laboratory Practice.
aVehicle—polyethylene glycol 400.
bVehicle—0.5% (w/w) hydroxypropylmethylcellulose and 0.1% (w/w) Tween 80.
cBecause of emesis, diarrhea, and anorexia, the high dose of 250 mg/kg/day was discontinued on day 5 and the animals were given a 9-day drug holiday, after which the dose was reduced to 125 mg/kg/day and administered for 29 days.
dBecause of the absence of notable toxicity and following the results of the plasma analysis, the high dose was increased from 300 mg/kg/day to 500 mg/kg/day commencing on day 148 and 147, for males and females, respectively.
eResults of pancreatic findings.
Statistical Analysis
Organ weights and organ-to-body weight ratios were analyzed using analysis of variance, followed by application of Dunnett’s (Dunnett 1955) or Williams’ (Williams 1971) multiple comparison procedure. For clinical chemistry data, nonparametric multiple comparison procedures were utilized. Shirley’s (Shirley 1977) or Dunn’s (Dunn 1964) test was utilized, based on an assessment of dose-related trend using Jonckheere’s test (Jonckheere 1954).
Results
Single-Dose Study in Rats and Mice
On study day 3 following a single oral dose of GI181771X, test article–related changes were limited to the pancreas and consisted of diffuse acinar cell necrosis and inflammation (characteristic of necrotizing pancreatitis) in rats given 2,000 mg/kg. There were no mortalities, and after a 15-day nontreatment period, the pancreatic changes were nearly resolved with the presence of well-differentiated acini and acinar cells with zymogen granules and minimal residual interstitial inflammation observed. In mice, the pancreatic changes were much more pronounced on day 3 than in rats, including three early deaths: one male and one female given 2,000 mg/kg and one female given 1,000 mg/kg were found dead on day 5, day 7, and day 6, respectively. All three dose groups (500, 1,000, and 2,000 mg/kg) had similar pancreatic findings. On day 15, surviving mice had varying degrees of acinar cell regeneration characterized by well-differentiated acinar cells with abundant eosinophilic zymogen granules. However, the pancreatic changes were not fully resolved in mice on day 15 (Figure 1). Compared with mice, pancreatic changes were nearly resolved in rats 15 days after administration of a single dose.
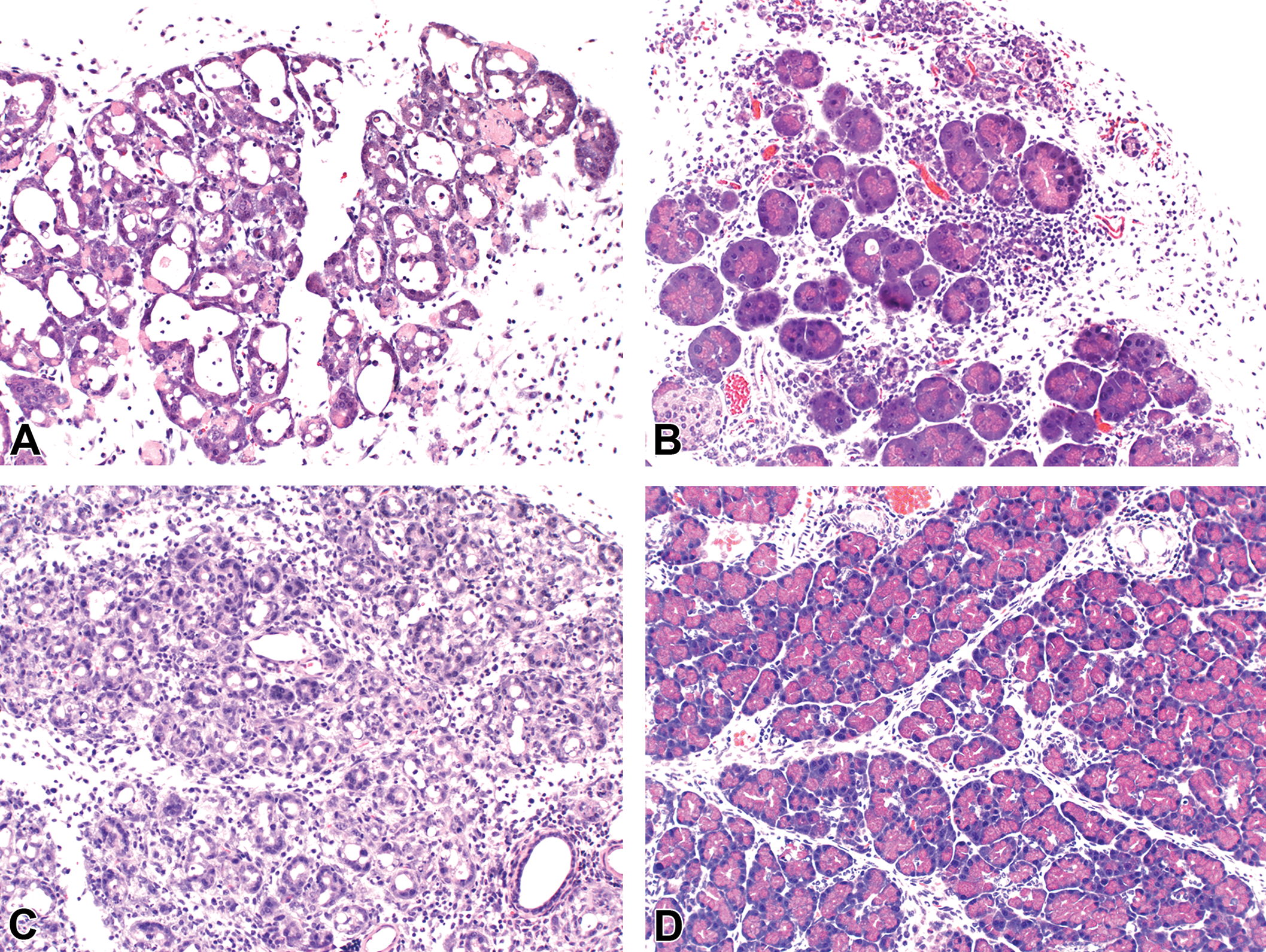
Acute study in mice and rats. A, Pancreas from a mouse 3 days after single oral dose of 2,000 mg/kg 771. Note diffuse necrotizing pancreatitis (acinar cell necrosis, zymogen granule loss, interstitial edema, inflammation, and dilated acini). H&E 200×. B, Pancreas from a mouse 15 days after single oral dose of 2,000 mg/kg 771 showing incomplete resolution. H&E 200×. C, Pancreas from a rat 3 days after single oral dose of 2,000 mg/kg 771. Note diffuse necrotizing pancreatitis with acinar cell regeneration. H&E 200×. D, Pancreas from a rat 15 days after single oral dose of 2,000 mg/kg 771. Note remarkable resolution (well-differentiated acini and acinar cells with zymogen granules); some residual interstitial inflammation. H&E 200×.
Repeat-Dose Rat Studies: 7-Day, 4-Week, and 26-Week
Amylase and Lipase
In the 7-day study, serum amylase activity was significantly decreased in animals given GI181771X at all doses. Similarly, a significant decrease in serum amylase activity was noted in the 4-week study but only in males given the high dose of 50 mg/kg/day and was reversible after cessation of treatment for 2 weeks. In the 26-week study, a significant decrease in plasma amylase activity was noted in males and females at ≥15 mg/kg/day. Plasma amylase activity remained lower in both males and females following the 4-week nontreatment period. In contrast to amylase, serum lipase activity was unchanged in both 7-day and 4-week studies. However, plasma lipase activity was minimally increased in the 26-week study in both males and females at all doses (Tables 4–6). However, this increase in plasma lipase activity was not observed after the 4-week recovery period following cessation of treatment.
The 7-day repeat-dose studies in Han Wistar rats—amylase and lipase activities.
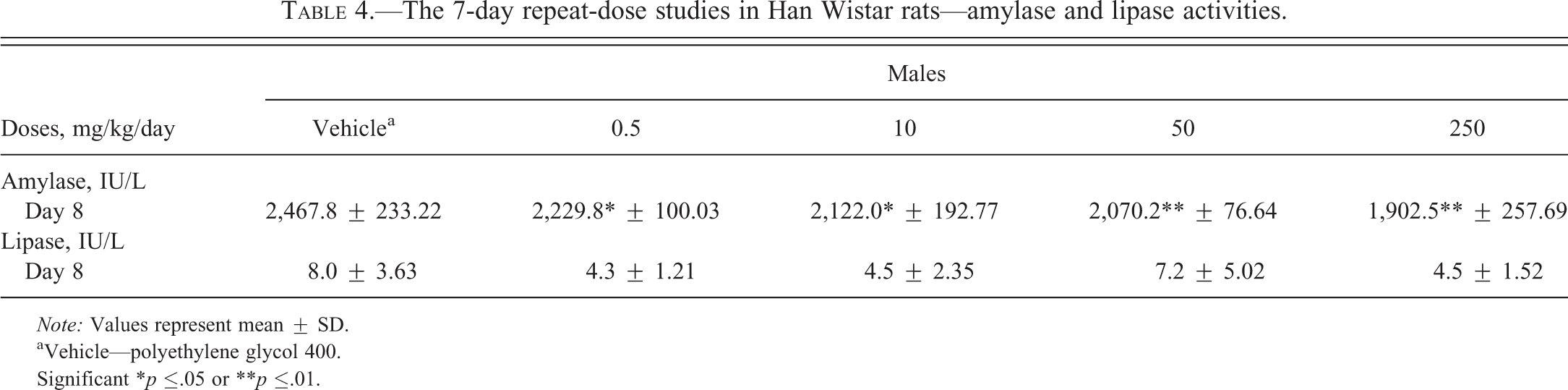
Note: Values represent mean ± SD.
aVehicle—polyethylene glycol 400.
Significant *p ≤.05 or **p ≤.01.
The 4-week repeat-dose studies in Han Wistar rats—amylase and lipase activities.
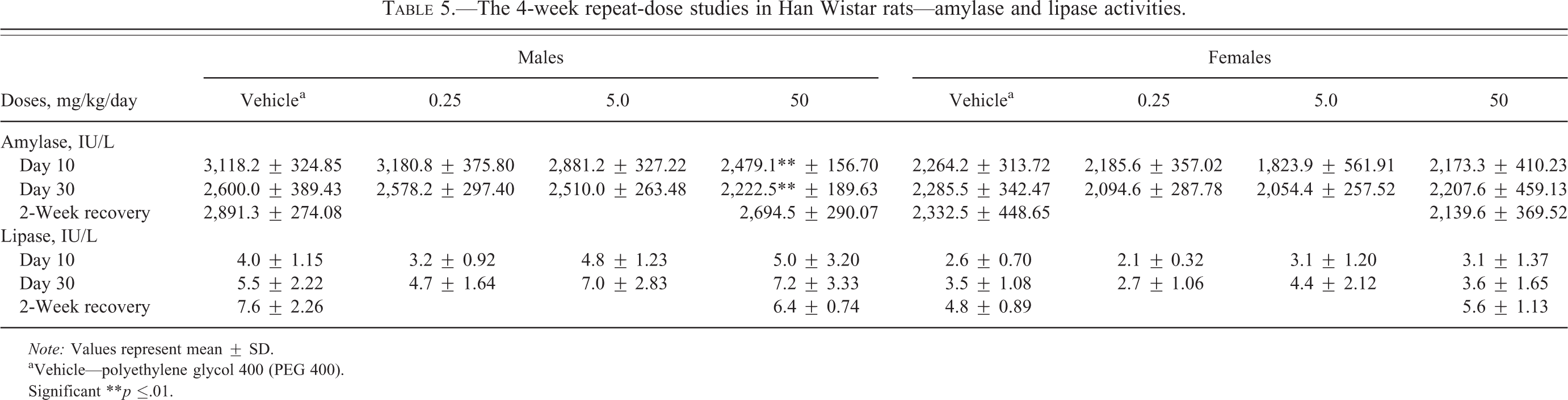
Note: Values represent mean ± SD.
aVehicle—polyethylene glycol 400 (PEG 400).
Significant **p ≤.01.
The 26-week repeat-dose studies in Han Wistar rats—amylase and lipase activities.
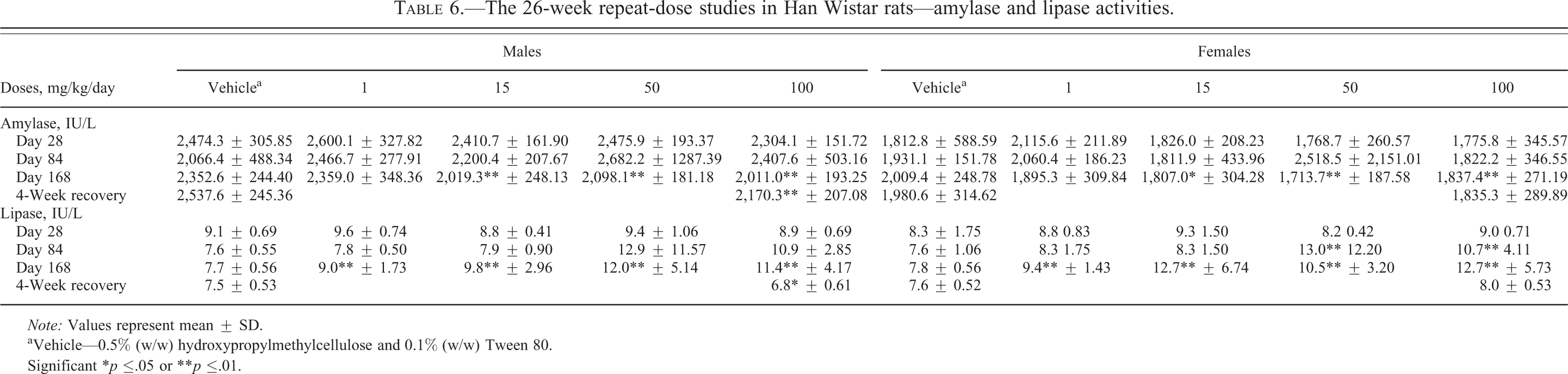
Note: Values represent mean ± SD.
aVehicle—0.5% (w/w) hydroxypropylmethylcellulose and 0.1% (w/w) Tween 80.
Significant *p ≤.05 or **p ≤.01.
Pancreatic Weights
In the 7-day study, a GI181771X-related biphasic response was noted in the pancreatic weights. Pancreatic weights (absolute and relative to body weight) were significantly increased in rats given 0.5 and 10 mg/kg/day GI181771X. In contrast, pancreatic weights were significantly decreased in rats given higher doses of GI181771X (50 and 250 mg/kg/day). In the 4-week and 26-week studies, pancreatic weights were significantly increased at all doses. However, increases were inversely related to the dose (i.e., lower doses had higher increases in pancreatic weights; Figure 2). Following the recovery period, weights were comparable with the controls.
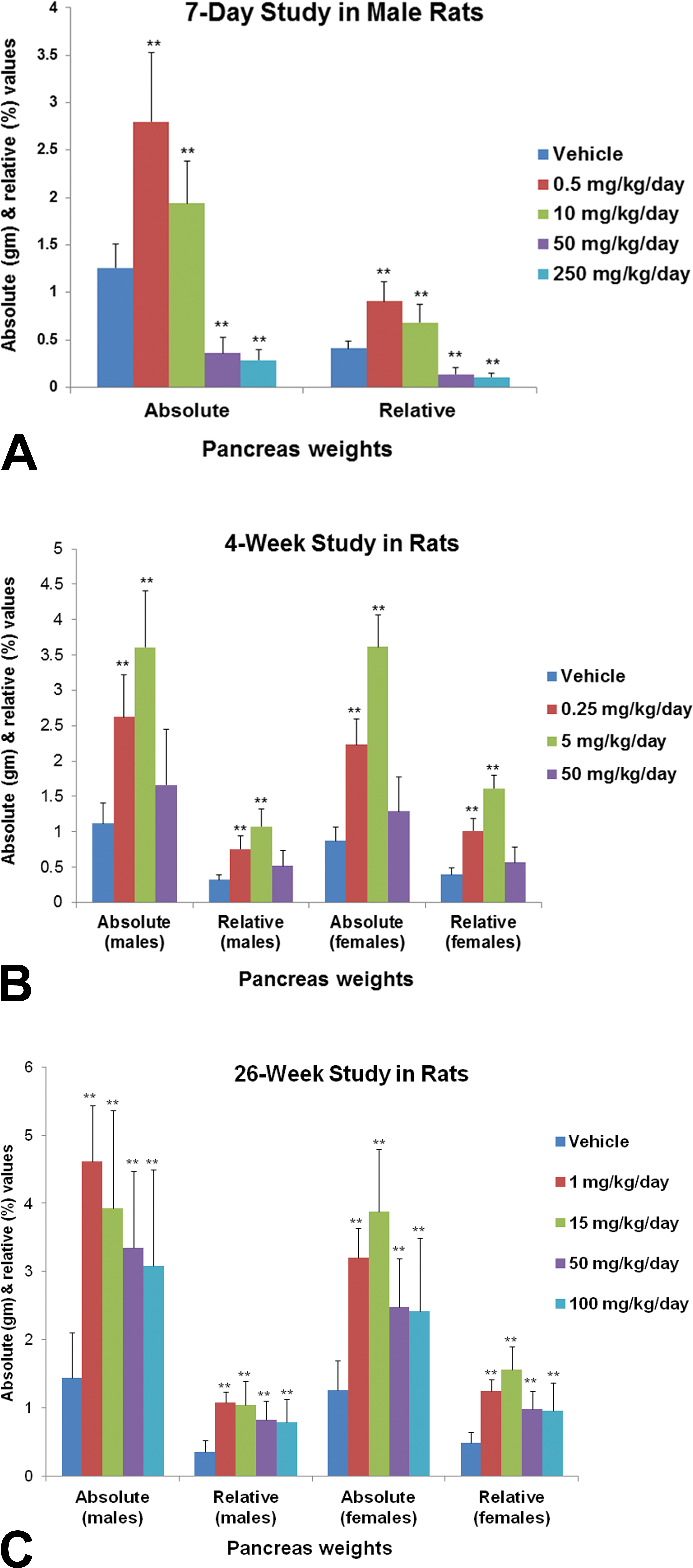
Repeat-dose rat studies. A, Biphasic response in absolute and relative pancreatic weights in 7-day rat study (increased at 0.5 and 10 mg/kg/day and decreased at 50 and 250 mg/kg/day). Note increased pancreatic weights in 4-week (B) and 26-week (C) rat studies. Data expressed as mean ± standard deviation. **Significant p <.01.
Macroscopic and Microscopic Findings
In the 7-day study, a biphasic response to GI181771X occurred in the pancreas of treated rats. Diffuse acinar cell hypertrophy, characterized by increase in cell size with or without increase in typical eosinophilic zymogen granules and cytoplasmic basophilia, was noted in animals given 0.5 and 10 mg/kg/day and correlated with enlarged and/or pale pancreas. These changes correlated with significant increases in absolute and relative pancreatic weights. In contrast, diffuse acinar cell atrophy (reduced cell size with relatively fewer zymogen granules and cytoplasmic basophilia) in animals given 50 or 250 mg/kg/day was associated with decreased pancreatic weight and size. Diffuse zymogen degranulation (hypertrophic or normal sized acinar cells with reduced or no intracytoplasmic zymogen granules) and multifocal interstitial inflammation (mostly mononuclear cell infiltrates) were noted in animals given 50 and 250 mg/kg/day.
In the 4-week study, macroscopic findings at the end of the treatment were limited to enlargement and/or pale coloration of the pancreas at all doses. GI181771X-related microscopic findings included varying degrees of diffuse acinar-cell hypertrophy and zymogen degranulation, multifocal acinar dilatation, and multifocal interstitial inflammation. The acinar cell hypertrophy was noted at all doses, but the morphologic details were variable among treatment groups. At 0.25 mg/kg/day, the acinar cell hypertrophy was manifested by enlarged cells with abundant zymogen granules and slightly enlarged nuclei and prominent nucleoli; however, at ≥5 mg/kg/day, this change was accompanied by varying degrees of zymogen degranulation. The hypertrophic acinar cells with zymogen degranulation had a slightly basophilic cytoplasm and noticeably enlarged nuclei and prominent nucleoli. The severity of acinar cell hypertrophy (based on the cell size) was greater in rats given 0.25 and 5 mg/kg/day than those given 50 mg/kg/day. The zymogen degranulation was noted in rats given 5 and 50 mg/kg/day, whereas multifocal interstitial inflammation (mostly mononuclear cell infiltrates) and acinar dilatation (dilated acini lined by flattened acinar cells) were noted in rats given 50 mg/kg/day. Following a 2-week recovery period in the 50 mg/kg/day dosed rats, multifocal interstitial mononuclear cell infiltration was still evident but reduced in severity.
In the 26-week study, enlargement and/or pale discoloration of the pancreas was seen at all doses at the end of the treatment period. Similar pancreatic changes described in the 7-day and 4-week studies were noted in the 26-week study. Significantly increased pancreatic weights at all doses were associated with diffuse acinar cell hypertrophy (Figure 3) with or without zymogen degranulation and cytoplasmic basophilia. Focal acinar cell atrophy (dilated acini lined by slightly flattened acinar cells) was noted at all doses in males and females given ≥15 mg/kg/day. Multifocal interstitial inflammation with or without fibrosis was noted at all doses in males and females given 50 and 100 mg/kg/day. Focal acinar cell hyperplasia (FAH) was observed in males at 15 mg/kg/day (3 of 16 males) and in females at 1, 15, and 50 mg/kg/day (1 of 11, 3 of 16, and 2 of 15 females, respectively; Figure 3) but not in both sexes at 100 mg/kg/day. FAH was manifested as a well-demarcated area (less than 3 mm in diameter) of tubuloacinar gland pattern with occasional mitotic figures and some degree of compression of adjacent parenchyma. These cellular characteristics along with the size of the affected area were consistent with FAH and not considered adenoma based on the Society of Toxicologic Pathologists (STP) nomenclature and diagnostic criteria (Hansen et al. 1995). Following the 4-week recovery period, multifocal interstitial inflammation/fibrosis was still evident but reduced. However, the morphology of the acinar cells was similar to that of controls.
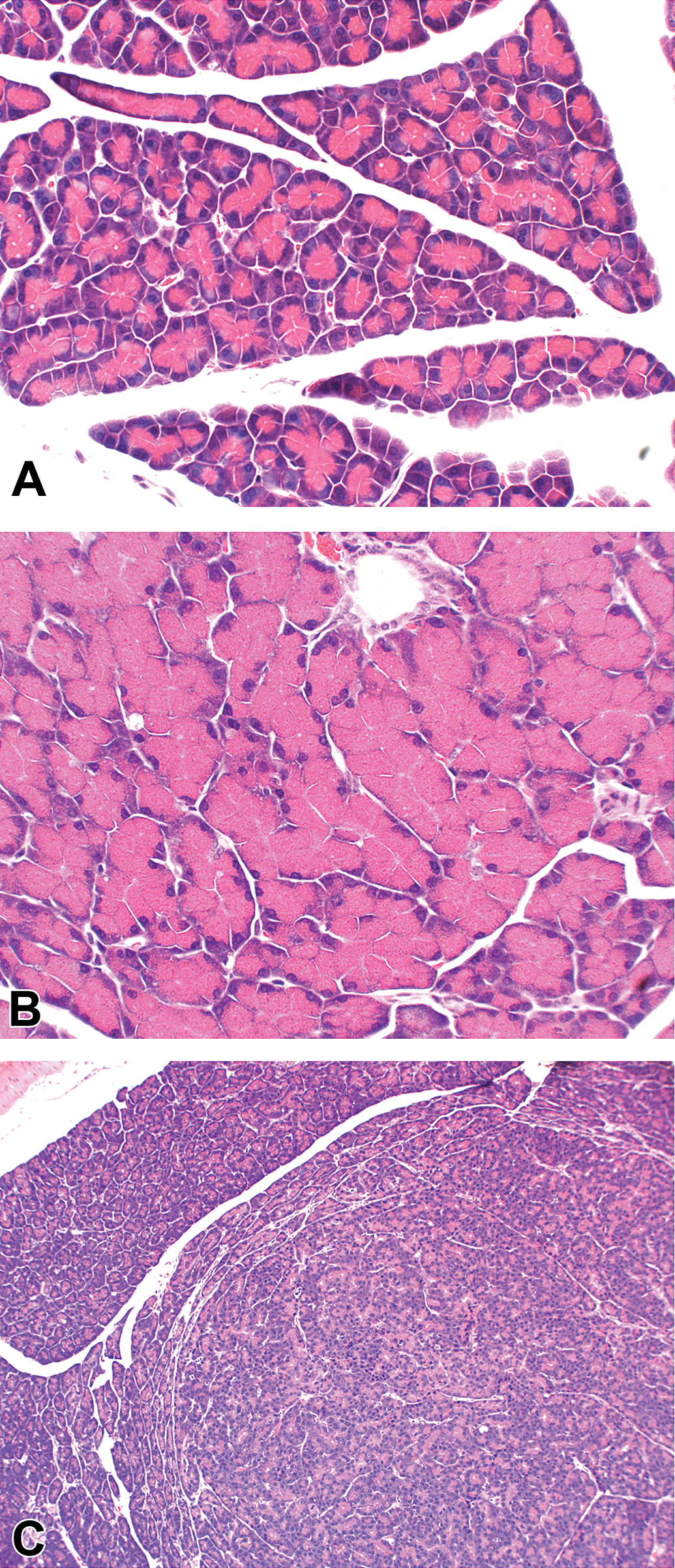
26-Week rat study. A, Pancreas from a vehicle (control) male rat. H&E 400×. B, Pancreas from a male rat given 1 mg/kg/day. Note diffuse acinar cell hypertrophy. H&E 400×. C, Pancreas from a male rat given 15 mg/kg/day. Note focal acinar cell hyperplasia. H&E 100×.
Repeat-Dose Monkey Studies: 30-Day, 26-Week, and 52-Week
In all repeat-dose studies in monkeys, no GI181771X-related effects were observed in amylase and lipase activities, pancreatic weights (absolute and relative to body weight), and pancreatic morphology (macroscopic and microscopic).
Discussion
Acute Single-Dose Study with GSK181771X in Mice and Rats
Our results showed pronounced species-related differences in the pancreatic responses caused by GI181771X, a selective CCK1R agonist. GI181771X-related necrotizing pancreatitis was noted in both mice and rats on study day 3 following a single oral dose of 2,000 mg/kg. However, the pancreatic changes (in particular, acinar cell necrosis and interstitial edema/inflammation) were much more pronounced in mice on day 3 than in rats, including unscheduled deaths at the high- (2,000 mg/kg group, one male mouse on day 5 and one female mouse on day 7) and mid-dose (1,000 mg/kg, one female mouse on day 6) groups. These differences were likely related to higher systemic exposure in mice (gender-averaged AUC of 8865 hr.ng/ml in mice vs. 4415 hr.ng/ml in rats for the 2,000 mg/kg dose ). Interestingly, there was remarkable exocrine cell regeneration and recovery (i.e., presence of well-differentiated acini and acinar cells with brightly stained zymogen granules) in both mice and rats after 15 days of nontreatment period. The exocrine cell recovery was more pronounced in rats than that of mice mainly because of differences in type and extent of cellular injury (i.e., acinar necrosis and inflammation were much more extensive in mice than in rats). Differences in receptor type between mice and rats may have also played a role in species-related susceptibility. Studies with the CCK analog, JMV-180, have shown that CCK receptors exist in two affinity states, such as low- and high-affinity CCK receptors. CCK-JMV-180 has been shown to act as a partial agonist in rats and a full agonist in mice resulting in species-dependent differences in biological activity (Ji, Kopin, and Logsdon 2000). Further study is required to understand the role of high- and low-affinity CCK receptors in GI181771X-induced pancreatitis in rodents. The histological features of GI181771X-induced pancreatitis were similar to CCK-8 or cerulein (CCK analog)-induced pancreatitis in rodents, a widely used experimental model of human pancreatitis (Longnecker and Wilson 2002; Green and Reeve 2008). CCK-8 or cerulein-induced acute pancreatitis is rapid and highly reproducible and is caused by exaggerated stimulation of CCK1R. Supraphysiological doses of CCK1R agonists caused severe acute pancreatitis in rodents, whereas OLETF rats (lacking CCK1R expression) developed less severe pancreatitis (Tachibana et al. 1997; Sato et al. 2003). The pathogenesis of necrotizing pancreatitis has been extensively evaluated and reported in the literature (see pathogenesis and molecular mechanisms).
Repeat-Dose Study with GI181771Z in Rats
In rats, GI181771X treatment caused wide-ranging morphological changes in the pancreas that were dose and duration dependent. These pancreatic changes included acinar cell hypertrophy, atrophy, zymogen degranulation, interstitial inflammation, and acinar cell hyperplasia. Although CCK-induced pancreatic changes have been well characterized in rodents, our findings highlight the dose- and/or duration-dependent aspects of the pancreatic changes observed in the longer duration studies of up to 26 weeks. In the 7-day repeat-dose study, pancreatic changes were distinctly dose dependent: diffuse acinar cell hypertrophy with increased number of zymogen granules at lower doses versus diffuse acinar atrophy with zymogen degranulation and interstitial inflammation at higher doses. Increased pancreatic weights (absolute and relative) at lower doses were correlated microscopically with diffuse acinar cell hypertrophy, while diffuse acinar cell atrophy contributed to the decreased pancreatic weights at higher doses. Similar dose-dependent changes have been described in rats given CCK-8 or cerulein, and these differences are likely caused by differences in receptor binding and activation (Hajri, Aprahamian, and Damge 1991; Trulsson at al. 2001). In contrast to the 7-day study, the dose-dependent pancreatic weight changes were not observed in 4-week and 26-week studies with GI181771X. In 4- and 26-week studies, pancreatic weights were increased in all treated groups, and these increases correlated with diffuse acinar hypertrophy. It is interesting to note that the pancreatic responses in rats given the same dose with comparable systemic exposure differed in longer duration studies (i.e., 4-week and 26-week) when compared with the 7-day study. For example, in rats given 50 mg/kg/day, diffuse acinar atrophy and decreased pancreatic weights were observed in the 7-day study. Rats given the same dose for longer periods (4-week and 26-week) had increased pancreatic weights and diffuse acinar cell hypertrophy, suggestive of an adaptive CCK1R response. Continuously administered CCK-8 causes downregulation of CCK1R in the rat pancreas (Ohlsson et al. 2000), however, the exact mechanism of these differences in the pancreatic response to 7-day versus 4-week or 26-week administration with GI181771X is unknown. Zymogen degranulation and interstitial inflammation were noted in all repeat-dose studies, however, these changes were dose related in 7-day and 4-week studies (i.e., noted only at higher doses) but not in the 26-week study (noted at all doses). As expected, FAH (well-demarcated area with some degree of compression of adjacent parenchyma) was observed in both males and females in 26-week rat study but not in shorter duration studies (7-day and 4-week). Acinar cell hypertrophy, zymogen degranulation, inflammation, and acinar cell hyperplasia have been described previously in CCK-induced pancreatic changes in rodents (Hajri, Aprahamian, and Damge 1991; De Dios et al. 1997; Trulsson et al. 2001). Despite varying degrees of GI181771X-related pancreatic changes, serum/plasma biomarkers, such as amylase and lipase activities, were not consistent among studies. In healthy rats, the parotid gland contributes more amylase to the circulation than the pancreas, and this end point has limited utility in monitoring pancreatic injury in this species (Nagy et al. 2001). Exposure to cerulein or CCK has been reported by others to cause transient increases in serum amylase and especially serum lipase activity consistent with the stimulation of pancreatic secretions (Tsukamoto et al. 1986; Kruse, Lasson, and Hage 1999). Decreases in amylase activity were noted in both 7-day and 26-week studies but not in the 4-week study (i.e., only high-dose males with significant decrease in amylase activity); this may reflect reduction in exocrine pancreatic secretory activity. Decreased food consumption may also lower serum amylase (Nagy et al. 2001). Similarly, an increase in lipase activity was noted only in 26-week studies at all doses but not in 7-day and 4-week studies.
Pathogenesis and Molecular Mechanisms of Pancreatic Findings
The pathogenesis and molecular mechanisms of CCK-induced pancreatic changes have been extensively evaluated and reported in the literature. Readers are encouraged to refer to these suggested review papers for details (Saluja et al. 2007; Mossner 2010; Sah, Garg, and Saluja 2012; Yu and Kim 2012). The pathologic cellular events are summarized below.
The Role of Pathological Basolateral Exocytosis
Several lines of evidence in CCK studies suggest that orderly secretion of zymogen at the apical pole is critical for physiological pancreatic acinar cell secretions. In supramaximal stimulation of CCK1R by CCK-8 or cerulein, acinar secretory activity increases dramatically, but membrane recruitment is insufficient relative to the strong demand of zymogen granule membrane, resulting in an inhibition of apical exocytosis, that is, granule fusion with the apical plasma membrane (Figure 4). The blockage of apical exocytosis leads to the misdirection of zymogen granules and basolateral exocytosis (i.e., release of granule content into the interstitial space; Gaisano and Gorelick 2009) and resultant pancreatitis. Although basolateral plasma membrane (BPM) accounts for 90% of acinar cell surface, BPM is not a typical site for normal physiological exocytosis. However, BPM has been shown to contain a complete set of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins (syntaxin 4 [Syn-4] and synaptosome-associated protein 23 [SNAP-23]) and cognate Munc 18c, suggesting that BPM has the membrane fusion machinery required for BPM exocytosis (Gaisano et al. 2001; Cosen-Binker et al. 2007; Lam et al. 2007). Recently, both apical and basolateral exocytotic events have been described in live acinar cells stimulated by maximal and supramaximal CCK concentrations (Fernandez, Liang, and Gaisano 2011). However, the significance of basolateral exocytosis alone to the pathogenesis of acute pancreatitis has not been fully elucidated.
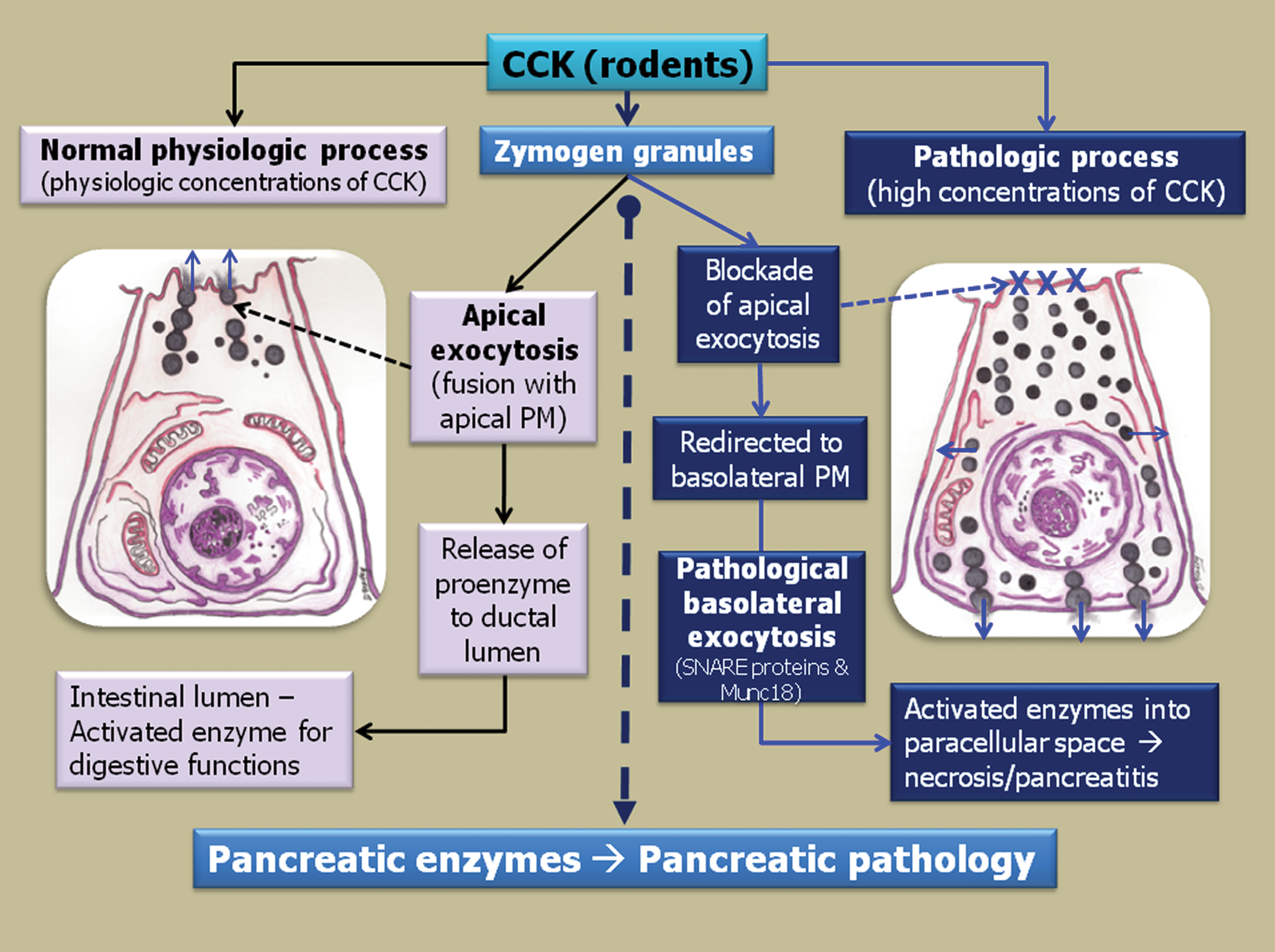
Pathological basolateral exocytosis (PBE) and pancreatic injury. In supramaximal stimulation of cholecystokinin 1 receptor (CCK1R) by CCK-8 or cerulein, acinar secretory activity increases dramatically, but membrane recruitment is insufficient relative to the strong demand of zymogen granule membrane, resulting in an inhibition of apical exocytosis (granule fusion with the apical plasma membrane). The blockage of apical exocytosis leads to the misdirection of zymogen granules and PBE (i.e., release of granule content into the interstitial space) and resultant pancreatitis. Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins and cognate Munc 18c are involved in PBE.
Roles of Cytosolic Calcium and Lysosomes in Zymogen (trypsinogen) Activation
In addition to the blockage of apical exocytosis and subsequent basolateral exocytosis, considerable evidence suggests that intracellular signaling (e.g., increased cytosolic calcium, activation of pancreatic proenzymes or zymogens) and extracellular events (e.g., dissociation of cellular contacts, inflammatory cell infiltration) play important roles in the pathophysiology of acute pancreatitis (Bialek et al. 1991; Luthen, Niederau, and Grendell 1995; Grady et al. 1996; Saluja et al. 2007; Muili et al. 2012). Figure 5 summarizes the molecular mechanism of zymogen activation, necrosis, inflammation, cellular growth (hypertrophy and hyperplasia), and neoplasia. The binding of CCK or CCK agonists to the CCK1R results in a conformational change in this receptor, which triggers an exchange of guanosine triphosphate (GTP) for bound guanosine diphosphate (GDP) at the G protein, with the dissociation of the GTP-bound α-subunit from its β and γ subunits. The GTP-bound α-subunit then activates phospholipase C (PLC), and PLC cleaves phosphatidylinositol 4, 5-bisphosphate to form inositol 3, 4, 5-triphosphate (IP3) and diacylglycerol (DAG; Trimble et al. 1987; Matozaki and Williams 1989). IP3 then moves to the endoplasmic reticulum and stimulates the release of calcium stores (Muallem et al. 1985). Along with the IP3 pathways, cyclic adenine dinucleotide phosphate (ADP) ribose (cADPr ), and nicotinic acid adenine dinucleotide phosphate (NAADP) have been implicated in the release of calcium from intracellular stores. Toxic Ca2+ signals generated by excessive liberation of Ca2+ from the endoplasmic reticulum activates calcineurin (Ca2+/calmodulin-activated phosphatase), which then mediates pathological intra-acinar cell zymogen activation (Husain et al. 2007; Shah et al. 2009; Muili et al. 2012) and subsequent autodigestive injury culminating as overt pancreatitis. Similarly, lysosomal cathepsin B has been shown to play a role in zymogen activation (Halangk et al. 2000; Saluja et al. 2007).
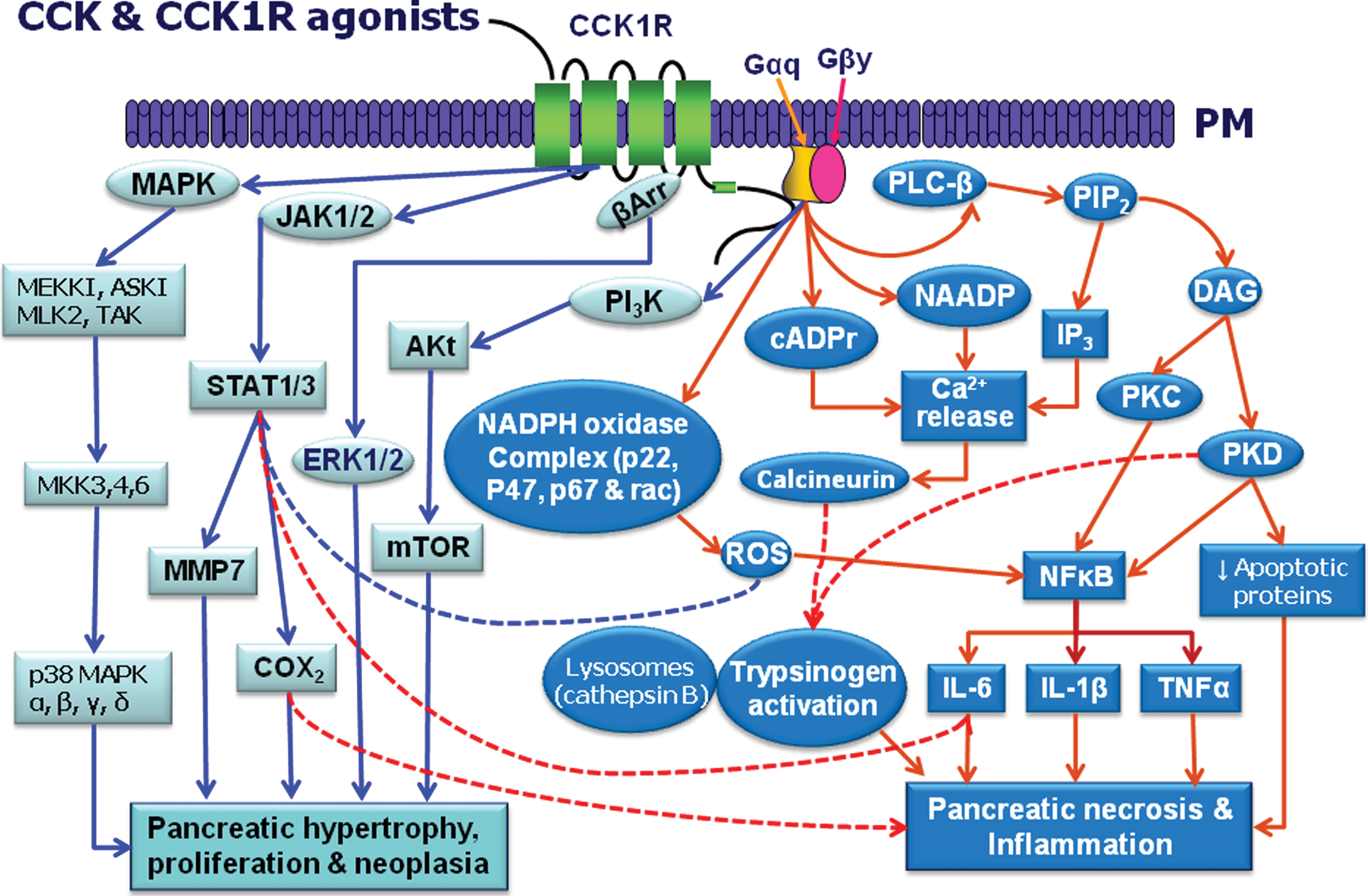
Schematic illustration of various signaling pathways involved in the pathogenesis of pancreatic responses (necrosis, inflammation, hypertrophy, hyperplasia, and neoplasia) by cholecystokinin (CCK) or CCK 1 receptor (CCK1R) agonists. Activation of CCK1R by CCK/CCK agonist causes activation of phospholipase C (PLC) with cleavage of phosphatidylinositol 4, 5-bisphosphate (PIP2)to form inositol 3, 4, 5-triphosphate (IP3) and diacylglycerol (DAG). IP3, nicotinic acid adenine dinucleotide phosphate (NAADP), and cyclic ADP-ribose (cADPr) pathways implicated in the release of Ca2+ and subsequent activation of zymogens through the activation of calcineurin; additionally, lysosomal cathepsin B has been shown to activate zymogens. DAG activates protein kinase C (PKC), protein kinase D (PKD), and nuclear factor-κB (NF-κB) and expression of interleukin 6 (IL-6), IL-1β, and tumor necrosis factor-α (TNF-α). PKD also inhibits apoptotic proteins to promote necrosis. CCK1R may activate nicotinamide adenine dinucleotide phosphate(NADPH) oxidase complex and produce reactive oxygen species (ROS), and ROS then activates NF-κB. CCK1R also activates Janus kinase (JAK 1/2) and signal transducers and activators of transcription (STAT 1/3), which induce the expression of inflammatory cytokines, metalloproteinase 7 (MMP-7), and cyclooxygenase 2 (COX-2). CCK or CCK1R agonist activates mitogen activated protein kinase (MAPK) pathways, such as extracellular-regulated kinases 1 and 2 (ERK1 and ERK2), p38 MAPK, and phosphatidylinositol 3-kinase (PI3K). PI3K then activates Akt, and Akt is known to have several targets including mammalian target of rapamycin (mTOR).
Roles of Protein Kinase C and D (PKC and PKD) Activation
DAG activated by PLC then activates PKC isoforms δ and ∊, downstream PKD, and nuclear factor-κB (NF-κB). Early intra-acinar NF-κB activation has been shown to play an important role in the pathogenesis of inflammatory response (Rakonczay et al. 2008; Sah, Garg, and Saluja 2012). NF-κB is a nuclear transcription factor responsible for regulating the transcription of a wide variety of genes involved in immunity and inflammation (interleukin [IL]-6, IL-1b, tumor necrosis factor-α [TNF-α]; Rakonczay et al. 2008; Yu and Kim 2012). Furthermore, the level of NF-κB activation correlated with the severity of pancreatitis in mice, and prolonged NF-κB activation resulted in chronic pancreatitis (Huang et al. 2013). Similarly, PKD has been shown to mediate NF-κB activation and inappropriate intracellular zymogen activation in pancreatitis (Yuan et al. 2008; Thrower et al. 2011). PKD activation has been shown to promote necrosis and inhibit apoptosis by inhibiting antiapoptotic proteins (X-linked inhibitors of apoptosis proteins, Fas-associated death domain protein-like IL-1beta-converting enzyme [FLICE]-inhibitory protein, and survivin; Yuan et al. 2012). The increased antiapoptotic proteins suppress caspase activation and apoptosis. The inhibition of caspases resulted in decreased cleavage/inactivation of receptor-interacting protein kinase 1 which, in turn, led to increased necrosis and severity of pancreatitis.
Roles of Janus Kinase (JAK), Phosphatidylinositol 3-kinase (PI3K), and Mitogen-Activated Protein Kinase (MAPK)
In addition to PKC and PKD, the binding of CCK or CCK agonists to the CCK1R also activates JAK (1/2) and signal transducers and activators of transcription (STAT 1/3), which induces the expression of inflammatory cytokines (IL-6, IL-1b, and TNF-α) and cancer-related genes such as metalloproteinase 7 (MMP-7) and cyclooxygenase 2 (COX-2) in pancreas. The activation of JAK1/2 and STAT 1/3 increases pancreatic growth and causes neoplasia. The binding of CCK or CCK agonists to the CCK1R may also activate nicotinamide adenine dinucleotide phosphate oxidase complex (Nox1, p67, p22, p47, a small G protein rac) and produce reactive oxygen species (ROS) which in turn activates JAK/STAT as well as NF-κB (reviewed in Yu and Kim 2012). Finally, the class I PI3K-signaling pathway plays many physiologic roles, including vesicle trafficking, cell growth, DNA synthesis, regulation of apoptosis, and cytoskeletal changes (Vanhaesebroeck et al. 2001). PI3K activated by CCK phosphorylate and activate Akt, and Akt is known to have several targets including mammalian target of rapamycin (mTOR; Vanhaesebroeck et al. 2001). mTOR activation controls protein synthesis, pancreatic growth (acinar cell hypertrophy), and proliferation (acinar cell hyperplasia; Bragado, Groblewski, and Williams 1997; Crozier et al. 2006; Borders, Bivona, and Medina 2010). Furthermore, the PI3K pathway has been implicated in the activation of the NF-kB transcription factor and intracellular activation of trypsinogen and resultant pancreatitis (Gukovsky et al. 2004). MAPK pathways, such as extracellular-regulated kinases 1 and 2 (ERK1 and ERK2) and p38 MAPK stimulated by CCK1R activation, have been shown to regulate a number of important cellular processes, including gene transcription, protein translation, metabolism, and cytoskeletal function. ERK1 and ERK2 play an important role in cell growth and mitogenic proliferation as well as the enhancement of cell cycle entry (Duan and Williams 1994; Robinson and Cobb 1997).
Species-Related Pancreatic Responses—Rodents, Cynomolgus Monkeys, and Humans
Repeat-dose studies in rats and monkeys and the 24-week clinical trial in overweight and obese patients with GI181771X showed convincing species differences in the pancreatic responses. The systemic exposures in the 26-week rat study ranged from 1,750 to 186,000 (AUC hr.pg/ml) for 1-, 15-, 50-, and 100-mg/kg/day dose groups; whereas in the 52-week monkey study, the systemic exposures were 15,500 to 312,000 (AUC hr.pg/ml) for 50, 125, and 300/500 mg/kg/day. Despite higher doses and systemic exposures of GI181771X in monkey compared with rat, and a longer treatment period (up to 52 weeks in monkeys compared with 26 weeks in rats), there were no test article–related effects on serum amylase, lipase activities, pancreatic weights (absolute and relative to body weight), and pancreatic morphology (macroscopic and microscopic) in monkeys. Similarly, the 24-week randomized double-blinded clinical trial with GI181771X in overweight and obese patients showed no laboratory, hepatobiliary, or pancreatic abnormalities (abdominal ultrasound and magnetic resonance imaging) (Jordan et al. 2008). In contrast, rats responded with varying degrees of pancreatic (morphologic) effects even at the lowest dose of 0.25 mg/kg/day GI181771X. These species-related differences are attributed to the differences in CCK1R expression/distribution and CCK responses. Previous reports had indicated that there are species differences in the expression of CCK receptor subtypes in the pancreas. The CCK1R expression has been difficult to demonstrate on human acinar cells because of either very low levels or nondetectable levels of this receptor. The quantitative reverse transcription–polymerase chain reaction (RT-PCR) technology demonstrated near absence of CCK-A receptor messenger RNA (mRNA) in human acinar cells (Ji et al. 2001). Furthermore, in-situ hybridization failed to detect CCK1R mRNA and location of CCK1R in human acinar cells (Ji et al. 2001, 2002). Galindo et al. (2005) have detected CCK1R transcription in human pancreas using advanced quantitative RT-PCR technology; however, the cell type expressing it was not determined. Like humans, CCK1R is expressed in low levels in brain and pancreatic acinar cells of cynomolgus monkeys (Holicky et al. 2001). In contrast, CCK1Rs are abundant on the rodent pancreatic acini, and CCK is a potent stimulant of pancreatic enzyme secretion in rodents (Wank, Pisegna, and de Weerth 1994; Monstein et al. 1996). Furthermore, human pancreatic acini express exclusively CCK2R, whereas rodent pancreatic acini express almost exclusively CCK1R (Wank, Pisegna, and de Weerth 1994; Zhou, Povoski, and Bell 1995; Monstein et al. 1996).
CCK1Rs are highly conserved among different animal species, with 95% identical and 98% similar between rats and mice. Human CCK1R was 98% identical to the cynomolgus monkey and 90% to the rat CCK1R (De Weerth et al. 1993; Holicky et al. 2001). Cynomolgus CCK1R is also functionally indistinguishable from the human CCK1R (Holicky et al. 2001). It has been shown that such sequence differences have important functional significance. For example, differences in two amino acid residues in rat and mouse CCK1Rs (Leu-43 and Ileu-50 in rat and Val-43 and Phe-50 in mouse) have caused completely opposite effects in MAP/ERK kinase kinase-mediated Jun activation (Ibarz et al. 2006). Whether CCK acts directly on human pancreatic acinar cells to modulate physiological digestive enzyme secretion is still controversial. It has been shown that CCK or secretin stimulation did not produce any functional responses or significant binding for CCK1R in isolated human pancreatic acinar cells (Ji et al. 2001, 2002; Miyasaka et al. 2002). However, these isolated human acinar cells become responsive to CCK stimulation after adenoviral-mediated transfection of either CCK1R or CCK2R gene (Ji et al. 2001). These data suggest that human pancreatic acinar cells lacked CCK1R gene and do not respond to CCK stimulation. In contrast, Murphy et al. (2008) demonstrated that CCK at physiological concentrations directly elicits cytosolic Ca2+ signaling, activates mitochondrial function, and stimulates enzyme secretion in isolated human pancreatic acinar cells. In humans, therefore, there is wider acceptance of the concept that CCK-induced pancreatic enzyme secretion is mediated by a cholinergic mechanism (Adler et al. 1991; Soudah et al. 1992). Substantial evidence also indicated that CCK1R is expressed in vagal afferent and intrapancreatic neurons and pancreatic stellate cells in humans and rodents (Owyang and Logsdon 2004; Phillips et al. 2010). For example, neurologic stimulation using acetylcholine or its long-acting analog carbacol causes robust physiological responses in both rodents and humans. In vivo human studies with highly specific CCK1R antagonist MK-329 demonstrated a significant inhibition of bile secretion with slight pancreatic enzyme secretion and delayed gastric emptying, indicating that CCK is not an essential mediator of postprandial pancreatic enzyme secretion in humans (Cantor et al. 1992). Thus, differences in the distribution of the CCK1R in rats, monkeys, and humans may explain the lack of any pancreatic effects in monkeys and patients treated with GI181771X.
In conclusion, administration of GI181771X, a novel CCK1R agonist, resulted in species-related differences in the pancreatic response. Acute (up to 2,000 mg/kg GI181771X, as a single dose) and repeat-dose studies in mice and/or rats (0.25–250 mg/kg/day for 7 days to 26 weeks) showed wide-ranging morphological changes in the pancreas that were dose and duration dependent, including necrotizing pancreatitis, acinar cell hypertrophy/atrophy, zymogen granule degranulation, acinar cell hyperplasia, and interstitial inflammation. In contrast to rodents, pancreatic changes were not observed in cynomolgus monkeys given GI181771X (1 to 500 mg/kg/day with higher systemic exposure than rats) for up to 52 weeks. Similarly, no GI181771X treatment-associated abnormalities in pancreatic structure were noted in a 24-week clinical trial with obese patients (body mass index [BMI] >30 or >27 kg/m2) as assessed by abdominal ultrasound or by MRI. Interspecies-related differences exist in the CCK1R distribution on the pancreatic acinar cells, and as a result, GI181771X administration responded differently among rodents, monkeys, and humans. Therefore, GI181771X-induced pancreatic changes in rodents are unlikely to occur in cynomolgus monkeys or humans, and rodent findings cannot be extrapolated to humans.
Footnotes
Acknowledgments
The authors thank Robert Greenhill, Dennie Frailey, and Nikki Slocum for their contributions, and Alan Stokes, Holly Jordan, Richard T. Miller, Roger H. Brown, Sundeep Chandra, and Rick Adler for their critical review of this article.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.