
Abstract
Sphingosine-1-phosphate (S1P) is a major bioactive phospholipid, which binds to and activates a family of five G-protein-coupled receptors designated as S1P 1 (S1P1) through S1P5. The S1P1 receptor subtype, expressed primarily on lymphocytes, is known to play a critical role in the regulation of lymphocyte trafficking. S1P1 inhibitors result in the inhibition of lymphoid cell trafficking and are of interest to treat various inflammatory conditions. In this study, we describe a gastric finding associated with oral gavage administration of a small molecule S1P1 inhibitor to Sprague-Dawley rats. Rats were administered an S1P1 inhibitor once daily for 4 weeks and necropsies were conducted at the end of the dosing phase, and clinical pathology and histopathologic examination were performed. Lymphopenia and changes in lymphoid tissues were noted and were consistent with the pharmacodynamic effects for S1P1 inhibitory action. Histopathologic examination of the stomach revealed atrophy and depletion of gastric parietal cells in the glandular portion of the stomach. There are no literature data to suggest that this gastric effect is related to S1P1 pharmacology. Therefore, the mechanism of the observed gastric lesion is likely chemotype mediated.
Introduction
Sphingosine-1-phosphate (S1P) is a lipid mediator needed for lymphocyte egress from both primary and secondary lymphoid tissues. S1P acts mainly by binding to and activating specific cell surface receptors. S1P is involved in various inflammatory and autoimmune disorders such as rheumatoid arthritis (RA) and transplant rejection (Goetzl and Rosen 2004; Hla 2003; Lee et al. 1999). S1P receptors belong to the class of G protein-coupled receptors that constitute five subtypes, S1P1–5. The S1P receptor subtype 1 (S1P1) is located primarily on lymphocytes (Sanchez and Hla 2004). Lymphocyte egress is thought to be mediated by the binding of S1P1 receptor on lymphocytes. RA is a systemic autoimmune disease characterized by synovial membrane hyperplasia and progressive joint destruction as well as infiltration of inflammatory cells including activated T cells (Choy and Panayi 2001). S1P and T cells play a crucial role in the pathogenesis of RA (Kitano et al. 2006).
A sphingosine analog called FTY720 is a potent immunosuppressive drug that binds to all S1P receptor subtypes and decreases peripheral lymphocytes. The immunosuppressant mechanism of FTY720 (2-amino-2-[2-(4-octylphenyl) ethyl]-1, 3-propane-diol hydrochloride) depends on reduction of circulating lymphocytes via binding to S1P receptors (Enosawa et al. 1996). Preclinical data clearly showed that rats pretreated with FTY720 have significantly lower arthritis incidence, delayed onset of arthritis, and suppressed disease activities, indicating that the FTY720 is effective in preventing the occurrence and development of collagen-induced arthritis (CIA) in preclinical RA model (Matsuura, Imayoshi, and Okumoto 2000; Wang et al. 2007).
The drug used in this toxicity study is a potent small molecule S1P1 receptor inhibitor intended for oral administration and for the treatment of RA. Oral administration of this drug to normal Sprague-Dawley rats caused a dose- and time-dependent reduction in circulating lymphocytes, consistent with the pharmacodynamic effects for an S1P1 inhibitor (Forrest et al. 2004; Mandala et al. 2002; Matloubian et al. 2004; Xie et al. 2003). In addition, the drug was efficacious in ameliorating inflammation in the rat CIA model.
Parietal cells are highly specialized cells within the glandular portion of the stomach. These cells are stimulated by gastrin, histamine, and acetylcholine and produce hydrochloric acid of the gastric juice and an intrinsic factor that binds vitamin B12. There are no known correlations between the S1P1 lipid mediator and parietal cells. The objective of this study was to evaluate the potential toxicity and toxicokinetics (TKs) of an S1P1 inhibitor drug when administered orally to rats once daily for 4 weeks. The focus of this article is on the histopathologic characterization and risk assessment of an unexpected stomach lesion of parietal cell atrophy and depletion.
Materials and Methods
Animals
Male and female Sprague-Dalwey (Crl:D(SD)) rats obtained from Charles River Laboratories (Portage, MI) were assigned to this toxicity study. Animals were 6 to 8 weeks old and weighed approximately 150 to 350 g at the start of dosing. Animals were individually housed in stainless steel cages. Standard procedures and conditions were applied for animal care, feeding, and maintenance of room, caging, and environment. Animals were fed Certified Rodent Diet (Harlan Teklad) ad libitum, and water was supplied ad libitum via an automatic watering system. Animals were fasted overnight prior to blood collection for clinical laboratory measurements at scheduled necropsy. This study was conducted in accordance with guidelines for animal welfare and U.S. Food Drug Administration Good Laboratory Practice (GLP) regulations. The procedures used in this study were reviewed and approved by the Institutional Animal Care and Use Committee. A potent inhibitor small molecule drug at the S1P 1 (S1P1) receptor was synthesized by Pfizer Worldwide Research and Development. Dosing suspensions were prepared approximately weekly in aqueous 0.5% methylcellulose.
Experimental Design
Toxicity main study animals (10/sex/group) and a separate satellite group TK study rats (4/sex/group) were administered the drug once daily via oral gavage for 4 weeks at doses of 10, 100, or 1,000 mg/kg/day. Because of the significant reduction in body weight and food consumption, animals administered the 1,000 mg/kg/day dose were not dosed on days 9 through 11 and dosing was resumed on day 12 at 300 mg/kg/day. Therefore, the high dose was 1,000/300 mg/kg/day. Control animals received vehicle alone, which included aqueous 0.5% methylcellulose.
Experimental Procedures
Animals were observed twice daily for clinical signs. Ophthalmic examinations were conducted once during the predose and once during the last 7 days of the dosing phase. Body weights were determined once prior to dosing on day 1 and weekly thereafter. Food consumption was determined weekly and at scheduled euthanasia. Hematological and serum chemistry parameters were evaluated on day 28 at scheduled euthanasia in animals designated for toxicologic assessment. Blood samples containing EDTA as an anticoagulant were analyzed for complete blood count (CBC). Serum was analyzed for glucose, cholesterol, bilirubin, total protein, albumin, globulin, albumin–globulin ratio, phosphorus, calcium, chloride, sodium, potassium, creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and urea nitrogen. Complete necropsies were performed on all animals designated for toxicologic assessment. Representative samples of the following tissues were collected at necropsy, fixed in 10% buffered formalin (except as noted), and processed for light microscopy: adrenals, bone marrow, brain, epididymides (Davidson’s fixative, Fisher Scientific, Pittsburgh, PA), esophagus, eyes (Davidson’s fixative), Harderian gland, heart, kidneys, large intestine, liver, lungs, bone, mammary gland, mesenteric lymph node, ovaries, pancreas, pituitary, prostate, sciatic nerve, seminal vesicles, skeletal muscle, skin, small intestine, spinal cord, spleen, stomach, submandibular salivary gland, testes (Davidson’s fixative), thymus, thyroid/parathyroid, tongue, trachea, urinary bladder, uterus, vagina, and gross lesions. All tissues were sectioned (4 µm thick), stained with hematoxylin and eosin (H&E), and evaluated microscopically.
The TKs
TK parameters were assessed on days 1 and 28 from rats designated for TK assessment. Blood samples (serum) were collected at approximately 1, 4, 8, and 24 hr after dosing. Following blood sampling, animals designated for TK assessment were euthanized and discarded. No other procedures were conducted on these animals. Serum samples were analyzed for S1P1 inhibitor drug concentration using a validated liquid chromatography (LC)/mass spectroscopy (MS)/MS method. TK parameters of maximum concentration (Cmax), time to Cmax (tmax), and area under the concentration–time curve (AUC) were determined.
Statistical Analysis
Statistical analysis was conducted on data from animals designated for toxicity assessment and was conducted on food consumption, organ weight, and quantitative clinical laboratory data. One-way analysis of variance (ANOVA) was used to analyze the data. If the ANOVA is significant, Dunnett’s t-test was used for pairwise comparison between treated and control groups.
Results
Clinical Observations
Moribundities occurred at 1,000 mg/kg/day dose. One toxicity female administered 1,000 mg/kg/day was euthanized moribund on day 11 of the dosing phase. This animal had drug-related significant reductions in body weight (15%) by day 11 of the dosing phase and reduced food consumption (61% compared with the mean consumption of controls in week 1). The animal’s clinical signs included thin appearance, red hair coat of the nares and front legs, and yellow hair coat in the perineal area. A definitive cause of moribundity could not be determined by clinical pathology or histopathologic examination. In addition, two TK females administered 1,000 mg/kg/day dose were euthanized moribund on days 7 and 11 of the dosing phase, respectively. One animal had hunched posture; decreased feces; red hair coat of the nares, front paws, and perioral area; and yellow hair coat in the perineal area. The other animal had hunched posture and decreased feces.
Drug-related clinical signs occurred in the 1,000/300 mg/kg/day toxicity group included thin appearance, clear oral discharge, decreased feces, rough hair coat, red hair coat of the nares and front legs, and yellow hair coat in the perineal area. After dose reduction to 300 mg/kg/day on day 12 of the dosing phase, the only clinical signs observed in the study were rough hair coat and decreased feces on day 15 of the dosing phase. No remarkable drug-related clinical signs were seen at 10 and 100 mg/kg/day over the entire course of the study.
Body Weight
Drug-related reductions in mean body weight and total mean body weight changes were observed in the 1,000/300 and 100 mg/kg/day dose groups when compared with respective control. On day 8 of the dosing phase, 1,000 mg/kg/day males and females (dose level for this group was reduced to 300 mg/kg/day on day 12 of the dosing phase) had 18% lower mean body weights compared with respective control. On day 29, compared with the control group, the 1,000/300 mg/kg/day and 100 mg/kg/day groups’ mean body weights were lower, that is, 9% to 13% and 8% to 10%, respectively.
Food Consumption
Mean food consumption was lower in a dose-dependent manner in all drug-dosed groups in week 1 of the dosing phase; food consumption was reduced by 42%, 17%, and 11% in males and 51%, 13%, and 11% in females dosed with 1,000, 100, and 10 mg/kg/day, respectively. By the end of dosing phase, no differences in food consumption were noted in any of the test article–treated groups.
Ophthalmic Examination
No drug-related ophthalmic effects were seen.
Clinical Pathology
Drug-related decreases in white blood cells (WBCs), lymphocytes, basophils, and eosinophils were observed in all drug-dosed rats. Decreases in WBCs (0.19–0.43× control mean), lymphocyte (0.09–0.20× control mean) counts, eosinophils (0.44–0.56× control mean), and basophils (0–0.5× control mean) were noted in all drug-dosed animals at ≥10 mg/kg/day. The decrease in WBCs was mostly due to decreased lymphocyte counts (Figure 1); an expected primary pharmacologic effect of the drug. A partial contribution of stress to the effects on lymphocyte and eosinophil counts could not be totally ruled out.
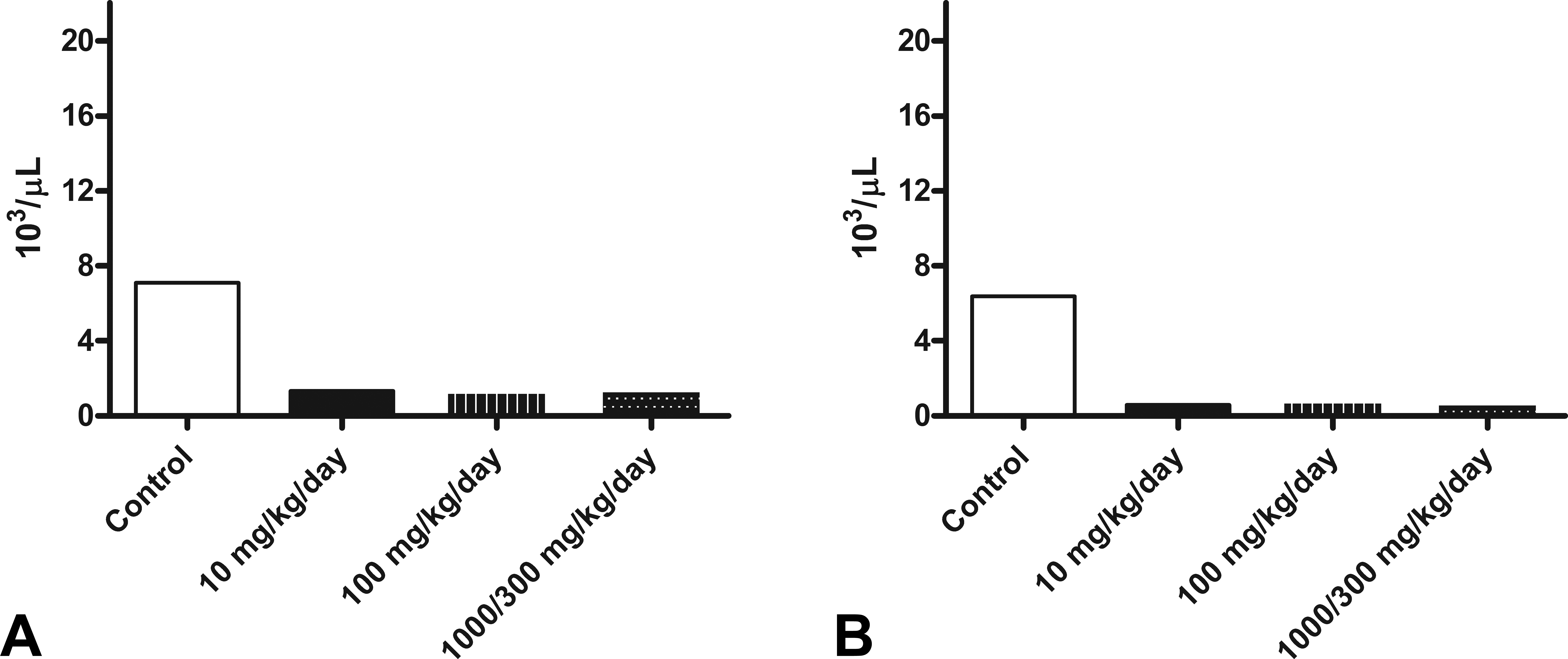
Mean lymphocyte counts. Note significant reduction in lymphocyte counts in the S1P1-agonist-treated male (A) and female (B) groups. S1P1 = Sphingosine-1-phosphate 1.
Minimally lower globulin (0.79–0.82× control mean) resulting in lower total protein (0.92–0.95× control mean) and higher albumin–globulin ratio (1.16–1.28× control mean) was noted in males and females at ≥10 mg/kg/day.
Minimal decreases in red blood cell (RBC) mass parameters (RBC, hemoglobin, and hematocrit) were noted in males at ≥100 mg/kg/day and in females at ≥10 mg/kg/day. These findings were associated with mild increases in reticulocyte counts and red cell distribution width in males at ≥100 mg/kg/day and in females at ≥10 mg/kg/day. Decrease in RBC mass could be due to decreased food consumption and body weight. Slightly higher platelet count was noted only in females, but not males, at ≥100 mg/kg/day.
The TKs
Serum samples of all surviving TK animals were analyzed. No apparent sex-related differences in exposure were noted and systemic exposure (assessed by Cmax and AUC0–24) increased with increasing dose.
Anatomic Pathology
Moribundities and Gross Pathology
No drug-related gross pathology findings were noted at necropsy. One female from the 1,000/300 mg/kg/day toxicity group and two females from the 1,000 mg/kg/day TK group were euthanized and no gross lesions were noted. All other toxicity animals survived until the scheduled terminal euthanasia.
Histopathology
Minimal to moderate parietal cell depletion and atrophy of the glandular stomach was noted in all males and females at ≥100 mg/kg/day (Figure 2). The stomach lesion was characterized by a diffuse decrease in the number and size of mainly the parietal cell component of the fundic region of the glandular stomach. Parietal cells within gastric glands were either unapparent or greatly reduced in size due largely to a decrease in cytoplasmic volume. The reduced size of the parietal cells gave the appearance of increased cellularity to the mucosal lining. Apoptotic cells, presumably parietal cells, were occasionally observed. This parietal cell depletion and atrophy was considered an adverse finding in males and females at ≥100 mg/kg/day.
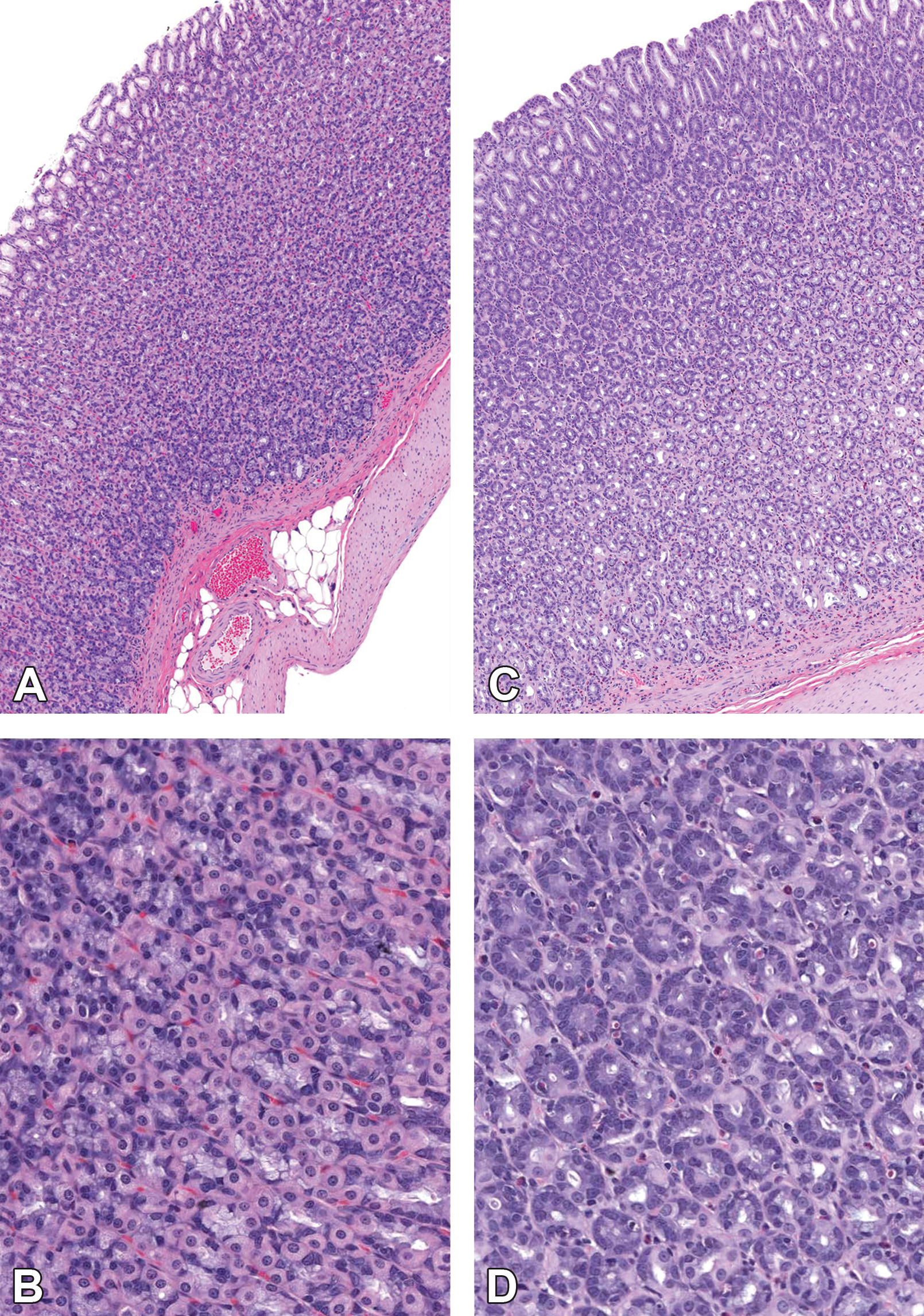
Rat stomach histopathology from control (A, B) and S1P1-inhibitor drug-dosed rats (C, D). Note the presence of abundant parietal cells in the normal glandular stomach (A, B) and parietal cell atrophy and depletion of the glandular stomach in the S1P1 drug inhibitor–dosed rats (C, D). H&E. S1P1 = Sphingosine-1-phosphate 1.
Drug-related and expected pharmacologic microscopic findings occurred in the lymphoid tissues (thymus, spleen, and mesenteric lymph node). In the thymus, increased lymphocyte cellularity in the medulla and relative decrease in cortical thickness were noted at ≥10 mg/kg/day. Increased cellularity in the medulla was characterized by expansion of the medulla due to the accumulation of lymphocytes.
In the spleen, decreased lymphocyte cellularity of the marginal zone/periarteriolar lymphatic sheath (PALS) region and increased lymphocyte cellularity of lymphoid follicles in the white pulp were seen at ≥10 mg/kg/day.
In the mesenteric lymph node, decreased lymphocyte cellularity of the medulla/subcapsular sinusoidal spaces was noted at ≥10 mg/kg/day.
Discussion
Parietal cells are highly specialized cells in the glandular portion of the stomach (Yao and Forte 2003). Their physiological stimuli include the parasympathetic mediator acetylcholine, histamine, and the peptide gastrin (Yao and Forte 2003). There are several parietal cell proteins that have been implicated in the cell activation process such as proton pump, trafficking and membrane recycling proteins, regulatory kinases, and cytoskeleton proteins. The role of S1P may play in parietal cells is not known.
The exact mechanism of the unexpected gastric lesion of parietal cell atrophy and depletion in rats in this toxicity GLP study using the S1P1 inhibitor drug is not known. One possible mechanism may be related to S1P1 target pharmacology. However, literature search revealed that S1P1 is not known to be specifically expressed by gastric parietal cells. In a separate experiment, we have investigated, using immunohistochemistry, S1P1 expression in rat and cynomolgus monkey stomach and found that parietal cells lack S1P1 expression. In addition, the parietal cell atrophy and depletion was seen at the mid and high dose only which does not support a pharmacology-based mechanism. Furthermore, no gastric lesions have been reported in S1P1 knockout mice. Finally, no gastric lesions were noted in another toxicity study using a distinct structural analogue than the one used in this toxicity study. Based on this information, we believe this finding is likely chemotype mediated.
We have observed a similar gastric lesion with the same drug in the 4-week GLP toxicity study in cynomolgus monkeys at all doses of 12, 25, and 100 mg/kg/day. Thus, while the no observed adverse effect level (NOAEL) for the stomach lesion is 10 mg/kg/day in rats, an NOAEL could not be assigned in monkeys. To make a risk assessment, we compared the sensitivity of rats and cynomolgus monkeys to the effects on parietal cells. This was done by examining the median histopathology scores for stomach parietal cell lesion in rats and monkeys in the 4-week toxicity studies against mean exposure to the S1P1 inhibitor drug expressed in terms of Caverage (AUC/24 hr) for free (unbound) drug at steady state (Figure 3). The severity of the effect of the drug on parietal cells in male and female rat and male and female monkey stomach correlated very well with drug exposure, data for rats and monkeys falling along the same dose–response relationship. The conclusion that rat and monkey are equally sensitive to the effects on parietal cells is further strengthened by observation that a close structural analogue for this drug also caused similar parietal cell changes in rats and monkeys, which correlated very well with drug exposure, with data for rats and monkeys falling along the same dose–response relationship. Thus, rat and monkey share common dose–response curves for each drug, and exposure (AUC) at the NOAEL was similar in rat and monkey. Because rats and monkeys demonstrated the same sensitivity to the effects of the compound on parietal cells, it was appropriate to base the safety assessment for this finding on the NOAEL established for rats at 10 mg/kg/day in the 4-week toxicity study.
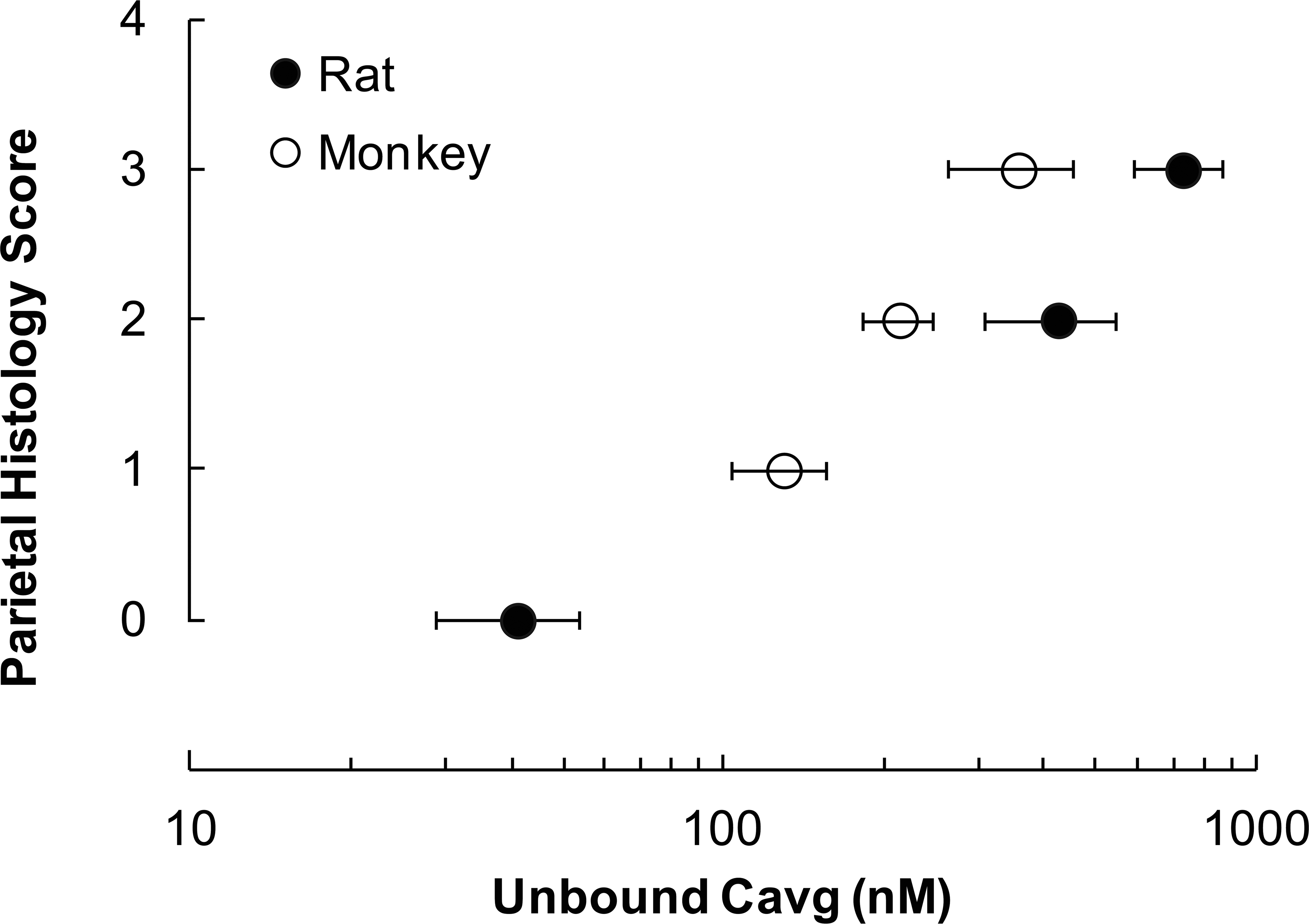
Median parietal cell atrophy and depletion histopathology score and mean ± SD free (unbound) Caverage (AUC/24 hr) at steady state in rats (closed dark circles) and monkeys (open circles) after S1P1 inhibitor once daily oral administration for 4 weeks. Note that the severity of the effect of the drug on parietal cells in rat and monkey correlates well with drug exposure, with data for rats and monkeys falling along the same dose–response relationship. S1P1 = Sphingosine-1-phosphate 1; AUC = area under the curve; SD = standard deviation.
Following oral administration to rats, the S1P1 inhibitor drug in our study caused dose-dependent decreases in lymphocyte counts (lymphopenia). Marked decreases in WBC counts at all dose levels, mainly accounted for by the decreases in lymphocytes, were noted. Lymphopenia is considered to be a pharmacodynamic biomarker for S1P1 inhibitor action. These observations are attributed to the pharmacology of S1P1 inhibitor drug where the S1P1 receptor subtype expressed on the surface of lymphocytes and is known to play a critical role in the regulation of lymphocyte recirculation, resulting in inhibition of lymphoid cell trafficking. Since S1P1 is essential for lymphocyte recirculation and can regulate lymphocyte egress from thymus and secondary lymphoid tissues (Brinkmann, Cyster, and Hla 2004; Lo et al. 2005; Matloubian et al. 2004), downregulated expression of S1P1 results in lymphocytes unable to respond to an obligatory egress signal provided by circulating S1P and subsequently unable to move out of lymphoid organs.
FTY720, a drug that binds to all S1P receptor subtypes, decreased the number of circulating blood lymphocytes and induced sequestration of circulating lymphocytes into the secondary lymphoid tissues and decreases lymphocyte infiltration into target organs (Chiba 2005). FTY720 prevented allograft rejection with high efficacy in animal models (Quesniaux et al. 1999) as well as in humans (Böhler et al. 2004; Budde et al. 2003), with reduction of CD4+ and CD8+ T cells. Additionally, peripheral decreases in lymphocytes were accompanied by microscopic findings in lymphoid tissues (spleen, thymus, and lymph nodes). Such pharmacological effects seen in lymphoid tissues with our compound are consistent with the pharmacologic activity of the drug. It is interesting to note that the increased lymphocyte cellularity in the medulla of the thymus noted by histopathologic examination agrees with studies by Wei et al. using two-photon microscopy to assess the pharmacologic effects of an S1P1 inhibitor, SEW2871. This S1P1 inhibitor has its strongest effect on medullary T-cell migration toward lymphatic sinuses (Wei et al. 2005). Approximately 5 min after SEW2871 application to the lymph node, the migration velocity of lymphocytes decreased from 6.5 μm/min to about 2 μm/min and washout of the S1P1 inhibitor restored migration velocity within 30 min. In contrast, T cells in the cortex migrated at a velocity of 10 to 12 μm/min and were completely unaffected by the presence of S1P1 inhibitor (Wei et al. 2005).
Collectively, our S1P1 inhibitor resulted in expected pharmacologic effects in 4-week toxicity studies and induced gastric parietal cell atrophy and depletion that is chemotype mediated. Risk assessment of stomach pathology finding in toxicity studies must consider dose–response relationship across species to accurately evaluate species sensitivity.
Footnotes
Acknowledgments
We thank Dr. Lawrence Wade for his help and Ashley Plaster for the help in preparing the images.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded and supported by Pfizer Worldwide R&D.