
Abstract
The colon serves as the habitat for trillions of microbes, which it must maintain, regulate, and sequester. This is managed by what is termed the mucosal barrier. The mucosal barrier separates the gut flora from the host tissues; regulates the absorption of water, electrolytes, minerals, and vitamins; and facilitates host–flora interactions. Colonic homeostasis depends on a complex interaction between the microflora and the mucosal epithelium, immune system, vasculature, stroma, and nervous system. Disruptions in the colonic microenvironment such as changes in microbial composition, epithelial cell function/proliferation/differentiation, mucus production/makeup, immune function, diet, motility, or blood flow may have substantial local and systemic consequences. Understanding the complex activities of the colon in health and disease is important in drug development, as xenobiotics can impact all segments of the colon. Direct and indirect effects of pharmaceuticals on intestinal function can produce adverse findings in laboratory animals and humans and can negatively impact drug development. This review will discuss normal colon homeostasis with examples, where applicable, of xenobiotics that disrupt normal function.
Introduction
In the past, little attention had been given to the normal physiology of the colon, as it was regarded simply as a tube that primarily managed waste and water. Certainly, the colon is essential for the reabsorption of water, electrolytes, and vitamins as well as in the movement of solid waste distally for excretion. However, in the last several years, studies have elucidated the complexity of colonic homeostasis and the importance of the trillions of microbes that live therein (the microbiome; Deplancke and Gaskins 2001; Kinnebrew and Pamer 2012; Wells et al. 2011). The balance between the microbiome and host is managed by what is termed the mucosal barrier. It is this barrier that separates the gut flora from the host tissues. It is comprised of luminal mucus, colonic epithelial cells, stromal and vascular elements of the lamina propria, and the enteric immune system, which act in a coordinated fashion to manage intestinal microbes to maintain colonic and systemic health (Deplancke and Gaskins 2001; Johansson et al. 2011; Kim and Ho 2010; Kinnebrew and Pamer 2012; Kunzelmann and Mall 2002; Wells et al. 2011). Disruptions in any of these components may result in mucosal damage, inflammation, and systemic disease (Deplancke and Gaskins 2001; Johansson et al. 2011; Kim and Ho 2010; Kunzelmann and Mall 2002; Morgan et al. 2012; Roda et al. 2010; Van der Sluis et al. 2006; Velcich et al. 2002; Yang et al. 2008). The focus of this article is to briefly describe the elements of the colon with relevant examples of xenobiotics that have been associated with perturbations of this organ.
The Microbiome
The colon contains the largest number of microbes per unit volume of the entire intestine. The microbiome is essential to the normal establishment of the intestinal immune system and also appears to play a role in epithelial cell maturation and mucosal angiogenesis (Hooper 2004; Hooper and Gordon 2001; Hooper and Macpherson 2010; Hooper et al. 2003; Molloy, Bouladoux, and Belkaid 2012; Noval Rivas et al. 2013; Russell et al. 2012; Stappenbeck, Hooper, and Gordon 2002). Recently, the function and importance of the intestinal microbiome on systemic health have begun to be elucidated. For example, intestinal microbial composition plays an important part in inflammatory and metabolic diseases, including autoimmune disease, insulin resistance, and lipid metabolism (Beyan, Wen, and Leslie 2012; Burcelin, Garidou, and Pomié 2012; Molloy, Bouladoux, and Belkaid 2012). Disruptions in the microflora (dysbiosis) can be caused by xenobiotics such as antibiotics but also by disturbances in motility and changes in diet (Bohm, Siwiec, and Wo 2013; Carlisle et al. 2013; Day et al. 2013; Marie et al. 2009; Shanahan 2012; Walker et al. 2011). Alterations in the intestinal microflora can create a proinflammatory environment or allow the overgrowth of pathogens (e.g., Clostridium difficile; Loh and Blaut 2012; Morgan et al. 2012; Pant et al. 2013). Disturbances in the microbiome can also increase susceptibility to allergic diseases, such as atopy and asthma (Beyan, Wen, and Leslie 2012; Burcelin, Garidou, and Pomié 2012; Chassaing et al. 2012; Koeth et al. 2013; Marra et al. 2006; Molloy, Bouladoux, and Belkaid 2012; Prioult and Nagler-Anderson 2005; Qin et al. 2012; Russell et al. 2013; Tremaroli and Backhed 2012; Vrieze et al. 2012).
Motility, the Enteric Nervous System, and the Interstitial Cells of Cajal (ICC)
In the colon, as with the rest of the intestine, smooth muscle contractions are both propagating and nonpropagating (mixing) and result from coordinated smooth muscle contraction and relaxation. Intestinal contractions, including propulsive contractions, normally continue regardless of the presence of ingesta (Bortoff 1976; Donnelly et al. 2001; Huizinga et al. 2011). Colon motility is directed through coordinated interactions between the enteric nervous system (motor neurons, interneurons, and intrinsic sensory neurons), the autonomic nervous system, specialized cells termed the ICC, and hormones and proteins/peptides (Lake and Heuckeroth 2013; Nezami and Srinivasan 2010; Rhee, Pothoulakis, and Mayer 2009). The enteric nervous system is intrinsic to the gut and controls not only motility but also glandular secretion (Nezami and Srinivasan 2010; Rhee, Pothoulakis, and Mayer 2009). The colon has inherent motility that does not require outside influences; however, external influences (e.g., extrinsic neuronal stimulus, proteins, hormones, etc.) modulate motility positively or negatively (Rae et al. 1998; Sarna 2010).
The terminology used to describe intestinal motility is variable and somewhat confusing (Huizinga et al. 2011). The mixing and propagation of ingesta through the colon are primarily the result of three different types of contractions: (1) rhythmic propulsive ripples (RPRs; also known as rhythmic phasic contractions and rhythmic myogenic ripples), (2) tonic contractions, and (3) ultrapropulsive colonic contractions (peristaltic contractions) termed rhythmic propulsive motor complexes (RPMCs), colonic migrating motor complexes, or giant migrating complexes (Huizinga et al. 2011). RPRs are driven by slow waves originating from ICC. RPRs primarily function in mixing contents with some slow propulsion of material. While RPRs slowly move contents distally in the colon of dogs and humans, they have a lesser role in the mixing or propulsion of ingesta in the colon of rodents (Huizinga et al. 2011; Sarna 2006; Sarna and Shi 2006). RPMCs propel ingesta and fecal material quickly over long distances by means of proximal smooth muscle contraction and distal smooth muscle relaxation (peristalsis) and play a dominant role in mixing and propagation of ingesta in the rodent colon (Huizinga et al. 2011; Sarna and Shi 2006). Unlike the dog and human, which have 2 to 5 RPMCs/day, rodents have 20 to 40/hr (Huizinga et al. 2011; Sarna and Shi 2006). The high frequency of RPMCs in rodents explains the more frequent fecal excretion pattern and fecal size/shape as compared to dogs, humans, and nonhuman primates. Motor function in the dog and human colon is similar and therefore the dog may serve as a better comparator for colon motility changes in humans than the rodent. Given these differences in mechanisms of mixing and propulsion between the rodent and nonrodent species, motility disturbances in nonclinical toxicology studies may not translate between species.
The enteric ganglia are located in the submucosa (submucosal/Meissner plexi) and between the inner circular and outer longitudinal smooth muscle layers (myenteric/Auerbach’s plexi; Sarna and Shi 2006). Signaling from both the submucosal and the myenteric plexi are important in gut motility, although the myenteric plexi have a greater role in motility than the submucosal plexi, which have a greater role in colonic secretion and absorption (Kunze and Furness 1999; Nezami and Srinivasan 2010; Sarna and Shi 2006). The enteric nerve fibers connect with other neurons, smooth muscle cells, blood vessels, ICCs, mucosal epithelium, and enteroendocrine cells and synapse with extrinsic autonomic nerves outside of the intestine (serving a sensory function; Lake and Heuckeroth 2013).
The primary excitatory neurotransmitter expressed by motor neurons and interneurons in the gut is acetylcholine (Ach), which induces smooth muscle contraction. Other important excitatory neurotransmitters include substance P (SP) and neurokinin A, which, like Ach, directly bind receptors on smooth muscle cells. Release of these neurotransmitters can be driven by peptides such as calcitonin gene–related protein and serotonin (5-hydroxytryptamine [5-HT]). Ach and other agonists activate mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinases (ERKs1/2), and p38 MAPK to stimulate the contraction of intestinal smooth muscle (Hedges et al. 1998; Ihara et al. 2007, 2009). Examples of primary inhibitory neurotransmitters are nitric oxide (NO), vasoactive intestinal peptide (VIP), gamma amino butyric acid, and adenosine triphosphate (Hwang et al. 2011; Rae, Khoyi, and Keef 1998). Prostaglandins also appear to have an inhibitory function on colonic motility (Fornai et al. 2010; Lin, Sarna, and Shi 2012). Serotonin (5-HT) has numerous receptor subtypes that may have paradoxical functions on motility (Dickson, Heredia, and Smith 2010; Liu et al. 2011). Only 5-HT released from enteric neurons (rather than from enteroendocrine cells) appears to have an impact on motility (Dickson, Heredia, and Smith 2010; Liu et al. 2011). Loss of enteric neuronal function in any segment of the gut will result in dysmotility (Obermayr et al. 2013), and any drug that alters the expression or function of these neurotransmitters can have an untoward effect on intestinal motility.
The ICC are specialized c-kit-positive cells that closely interact with enteric nerve fibers and are thought to electrochemically unite the colonic submucosa with the myenteric plexi and smooth muscles (Beckett et al. 2005; Garcia-Lopez et al. 2009; Huizinga et al. 2011; Komuro 2006; Sanders and Ward 2006; Ward et al. 2002; Ward and Sanders 2006). These cells cannot be identified on routine hematoxylin and eosin sections and are generally identified using immunohistochemical techniques. Gastrointestinal stromal cell tumors (GIST) derive from these cells (Sircar et al. 1999). ICCs are present in all layers of the gut wall and are electrochemically linked by gap junctions (Olsson and Holmgren 2011). ICCs have synapse-like contact with the enteric nervous system (Huizinga, Zarate, and Farrugia 2009). Depending on their location, they can have a wide range of receptors for various neurotransmitters, including 5-HT, bomebesin, cholecystokinin, protein kinase A, protein kinase C, M2 and M3 muscarinic receptors, neurokinin subtypes, somatostatin, VIP, and others (Garcia-Lopez et al. 2009). In the colon, ICCs are generally divided into the ICC-SM (within the submucosa), ICC-CM (within the circular muscle layer), ICC-LM (within the longitudinal muscle layers), and ICC-MY (associated with the myenteric plexus aka ICC-MP; Komuro 2006; Liu et al. 2012). The ICC-SM have a pacemaker function in the colon associated with slow wave contractions, which is in contrast to that in the small intestine in which the ICC-MY have a greater role in the slow wave pacemaker activity (Bayguinov, Hennig, and Smith 2010). Data suggest that the ICC-MY may work in conjunction with the slow waves of the ICC-SM to couple the circular and longitudinal muscles during colon contractions. The role of ICC-SM as a slow wave pacemaker in the colon is consistent with the findings in mice with mutations in the dominant white spotting/c-kit mutation KitW/KitW-v (white spotting locus Wv; Huizinga et al. 1995). These mice have severely underdeveloped ICC-MY and, in addition to a myriad other problems, have severely decreased small intestinal contractility with otherwise relatively normal colonic contractility. In mice, the density of ICC (particularly ICC-MY) decreases from the proximal colon to the rectum, which is in contrast to humans who have the highest ICC density in the transverse colon and lower densities in the other portions of the colon (Hagger et al. 1998). A wide assortment of maladies, including diabetes, has been associated with reductions in the numbers of the various ICC in the intestine, and this reduction is believed to be a cause of constipation in these diseases. It is thought that these reductions in ICC are secondary to the ailments (Becheanu et al. 2008; Der et al. 2000; He et al. 2001; Porcher et al. 2002; Ro et al. 2010; Southwell 2008; Vanderwinden et al. 1996; Yamamoto et al. 2008).
Gut Motility and Xenobiotics
Normal gut motility can be adversely impacted by a variety of xenobiotics (Brock et al. 2012; Geboes, Hertogh, and Ectors 2006; Sarna 2006; Zeino, Sisson, and Bjarnason 2010). The most common colonic motility issues associated with drug side effects and human disease are those causing altered RPMCs. Slowed RPMCs allow for increased water absorption and may result in dry, hard feces and constipation, and abdominal discomfort (Cappell 2004; Zeino, Sisson, and Bjarnason 2010), while increased RPMCs result in diarrhea (Sarna 2010). Examples of xenobiotics associated with motility dysfunction are listed in Table 1. Because many therapeutics have additional receptor activation or blockade, motility-associated off-target functions may be manifested particularly at high doses.
Examples of common xenobiotics associated with dysmotility.

The Mucosal Barrier
The colonic mucosal barrier separates the microbiome from the tissues of the host. This barrier is comprised of mucus, colonocytes, immune cells, stroma, and the vasculature. Colonic homeostasis requires a fine balance between all of the components of the mucosal barrier.
The Mucus
Mucins are glycosylated proteins that are highly conserved between species. Their secretion is constitutive but can be induced by various stimuli (Kim and Ho 2010). Mucin composition varies depending on the location in the GI tract, and mucins can be differentially glycosylated even between adjacent crypts (Holmen-Larsson et al. 2013; Karlsson et al. 1997; Matsuo et al. 1997; Matsuoka, Pascall, and Brown 1999; McGuckin et al. 2011). There is a wide assortment of mucins, some of which are secreted and others are membrane associated (Kim and Ho 2010). Membrane-associated mucins are localized to the apical membrane and in part regulate epithelial cell inflammatory responses to pathogens (Kim and Ho 2010; Nishida et al. 2012). Disruptions in normal membrane-bound or secreted mucus production and glycosylation can result in decreased resistance to pathogens, increased intestinal inflammation, and increased risk of intestinal cancer (Heazlewood et al. 2008; Kim and Ho 2010; Luu et al. 2010; Moehle et al. 2006; Nishida et al. 2012; Resta-Lenert et al. 2011; Senapati et al. 2010; Van der Sluis et al. 2006; Velcich et al. 2002). For example, polymorphisms in the membrane-bound mucin MUC1 have recently been associated with the Crohn’s disease (Franke et al. 2010), a chronic inflammatory intestinal disease that appears to be driven at least in part by defects in innate immunity to intestinal microbes (McGuckin et al. 2011).
The primary secreted mucin in the colon is MUC2 but MUC5AC and MUC6 are also secreted (Johansson et al. 2011; Kim and Ho 2010). The secreted mucin layers are not evident in formalin-fixed samples but can be preserved using Carnoy’s fixative (Figure 1). Secreted mucus in the colon forms two distinct layers: an outer loose layer that contains bacteria and an inner dense layer adjacent to the mucosa, which is essentially sterile (Johansson et al. 2011). The outer loose mucin layer undergoes constant turnover, as mucus is degraded by bacteria and local enzymes and propagated along the intestinal tract. The outer mucous layer lubricates the passage of ingesta and protects the epithelium from physical and chemical trauma (Johansson et al. 2011). The inner dense mucous layer serves as both a physical and a chemical barrier between the microbial population and the intestinal epithelial cells (IECs). The inner mucous layer is a very dense gel that contains antimicrobial peptides, enzymes, and enteric organism-specific secretory immunoglobulin A (IgA), IgM, and IgG that reach the mucous layer by trancytosis (Mostov 1994) and are present in decreasing concentration from the epithelial cell membrane to the lumen (Johansson et al. 2011). There is some evidence that disruption in the expression of mucin-associated antimicrobial peptides such as β-defensins (O’Neil et al. 1999), thioredoxin, and peptidoglycan recognition proteins 1, 3, and 4 can alter the microbiota and may have a role in the development of Crohn’s disease and inflammatory bowel disease (IBD; Duerr and Hornef 2012; Kim and Ho 2010; Moehle et al. 2006; Wehkamp et al. 2005). Epithelial cells, including goblet cells, are responsible for the production of many of these factors. Expression and secretion of antimicrobial peptides are driven in a large part by the activation of the pattern recognition receptors (PRRs) of the innate immune system by intestinal microflora (Kinnebrew and Pamer 2012; McGuckin et al. 2011; Plaisancie et al. 2006). PRRs will be further discussed in the section on colonic immunity.
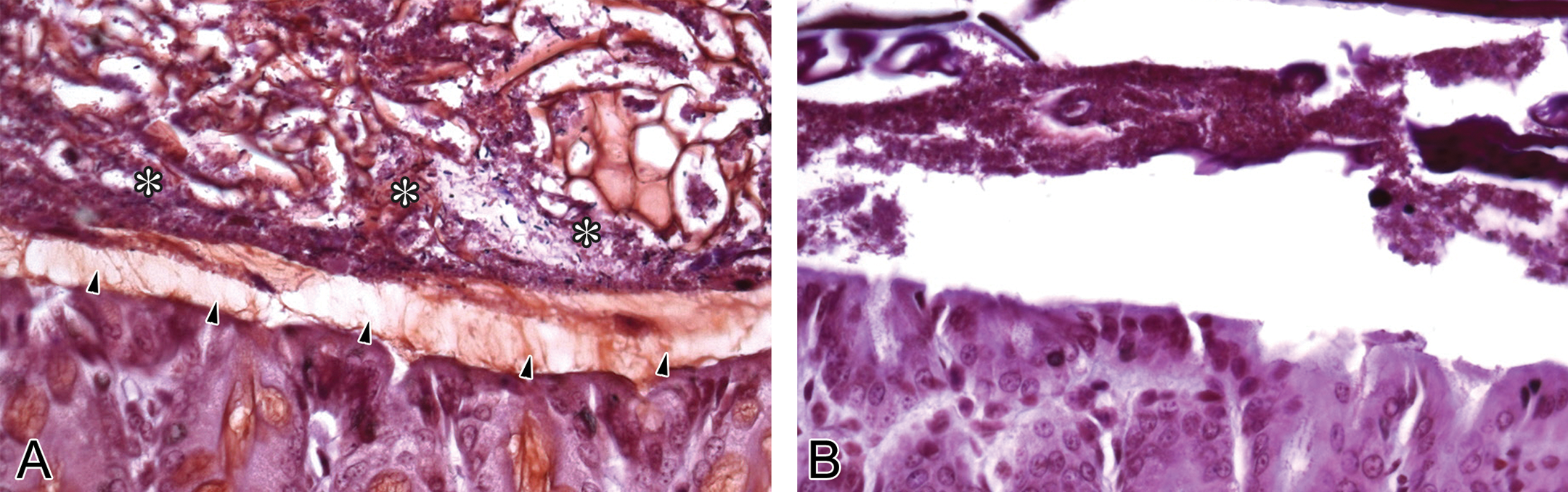
High-magnification photomicrographs depicting the dense inner (arrows) and loose outer (asterisks) mucous layers in the colon of the mouse. Gram’s stained control mouse depicting normal mucous layers (A) and the absence of mucous layers in a Muc2 mutant mouse (B).
The Epithelium: Stem Cell Niche
Within the colonic epithelium are three basic mature cell types: columnar cells, goblet cells, and enteroendocrine cells. Epithelial cell division occurs in the crypts, and differentiation increases as the cells migrate from the crypt to the surface where they are shed, resulting in constant replacement of the epithelium at the surface. Colonic stem cells are located at the base of the crypts (termed crypt base columnar cells or CBC cells; Barker et al. 2007; Simons and Clevers 2011). The CBC cells express leucine-rich repeat-containing G-protein-coupled receptors (LGRs)-4, -5, and -6 and are typically identified by their expression of LGR5 (Wright 2012). Stem cells generate transit amplifying (TA) cells that are the primary proliferative cell of the colon epithelium and differentiate into all absorptive and secretory epithelial cells in the colon (Munoz et al. 2012).
Stem cell proliferation and survival in the colon are dependent on Notch and Wnt signaling (Guruharsha, Kankel, and Artavanis-Tsakonas 2012; Haegebarth and Clevers 2009; Huch et al. 2013; Nakamura, Tsuchiya, and Watanabe 2007; Pinto and Clevers 2005a, 2005b; Riccio et al. 2008; van Es et al. 2005). Wnt and Notch signaling drive stem cell proliferation, and Notch receptor signaling is essential for stem cell maintenance and in guiding cell fate (Guruharsha, Kankel, and Artavanis-Tsakonas 2012; Haegebarth and Clevers 2009; Huch et al. 2013; Pinto and Clevers 2005a, 2005b; Riccio et al. 2008; van Es et al. 2005). Blocking Notch signaling reduces colonic crypt proliferation and increases LGR5+ epithelial cell apoptosis (Riccio et al. 2008; Rothenberg et al. 2012). Important sources of the ligands supporting LGR5+ stem cells (e.g., Wnt3, Notch ligands, EGF, and transforming growth factor [TGF]-α) in the small intestine are from Paneth and stromal cells (Clevers and Bevins 2013; Farin, van Es, and Clevers 2012; Kim, Escudero, and Shivdasani 2012; Medema and Vermeulen 2011; Sato et al. 2011). Unlike the small intestine, however, the colon generally lacks Paneth cells (although these may occasionally be present in the cecum and appendix of humans and in the colon of Apcmin mutant mice; Andreu et al. 2008). The source of LGR5+ stem cell supporting factors in the colonic epithelium has remained undetermined, but a recent study suggests that within the colon of the mouse are Paneth-like cells that may help sustain the LGR5+ stem cell population (Rothenberg et al. 2012). These are a goblet cell subpopulation which are cKit/CD117+ and also express the Notch ligands Dll1, Dll4, and epidermal growth factor (EGF), similar to Paneth cells (Rothenberg et al. 2012). These cKit+ cells (unrelated to ICC) are closely associated with the LGR5+ stem cells and appear to be the Paneth cell equivalent in the colon of the mouse. It is hypothesized that these Paneth-like cells are also present in the human colon (Rothenberg et al. 2012).
Canonical Wnt signaling results in the freeing of adenomatous polyposis coli (APC) tumor suppressor protein from beta-catenin, which translocates to the nucleus to activate proliferation. Mutations in APC gene Apc or beta-catenin have been associated with a number of cancers, in particular colon cancer in humans (Bodmer et al. 1987). Genetically engineered mice with mutations in the Apc gene (Apcmin mice) develop small intestinal lesions ranging from epithelial dysplasia to carcinoma, with few colonic tumors (Fodde et al. 1994; Ward and Devor-Henneman 2004). In the presence of colonic inflammation, some Apcmin mouse models will develop colon tumors at a much higher frequency than those without colonic inflammation (Bodmer et al. 1987; Yang et al. 2008).
Notch signaling relies on cell–cell contact and can mediate inhibition between adjacent cells to prevent identical differentiation patterns (Noah and Shroyer 2013; Schonhoff, Giel-Moloney, and Leiter 2004). There are 4 known Notch receptors (1–4) and 6 well-defined Notch ligands (Guruharsha, Kankel, and Artavanis-Tsakonas 2012; Nakamura, Tsuchiya, and Watanabe 2007). Ligand binding to Notch results in the cleavage of the Notch intracytoplasmic domain (NICD) by gamma secretase. NICD translocates to the nucleus where it forms a complex with DNA binding and transcription factors to activate the target genes (Guruharsha, Kankel, and Artavanis-Tsakonas 2012; Nakamura, Tsuchiya, and Watanabe 2007; van Es et al. 2005). The effects of these ligands can have both redundant and contradictory effects. For an excellent review of Notch signaling in the intestine, please see Noah and Shroyer (2013).
In contrast to previous dogma that the secretion occurs in the crypts while the surface epithelium is absorptive, colonic crypt and surface epithelial cells both have secretory and absorptive functions. There is a wide assortment of ion channels and ion exchangers on the colonic epithelial cells (Geibel 2005). Secretion is important to keep the mucous layer well hydrated and allow for movement of the mucus out of the crypts and through the colon (Geibel 2005). Many xenobiotics have been associated with disruptions in colonic secretion and absorption. For a detailed review of fluid homeostasis in the intestine, please see the publication by Kunzelmann and Mall (2002).
The Epithelium: Goblet Cells
In the colon, mature goblet cells make up 16% to 20% of the colonic epithelium (Karam, 1999; Katz et al. 2002). Differentiation of colonic TA cells into goblet and other secretory cells requires a downregulation in Notch signaling that results in the upregulation of ATOH1 (MATH1) and BMP4 and downregulation of HES family genes (Noah and Shroyer 2013; van Es et al. 2012). Goblet cells are essential in the colon, as they produce antimicrobial peptides and mucus. Induction of mucin production and secretion and goblet cell proliferation is driven in a large part by the presence and type of gut microflora (Kim and Ho 2010). Gnotobiotic mice, for example, have few goblet cells and a notably thinner mucous layer in the colon than in conventionally housed mice. Introduction of intestinal flora into gnotobiotic mice increases the number of goblet cells and mucin production (Deplancke and Gaskins 2001).
In addition to their important function in secreting mucus and antimicrobial factors, goblet cells have a close association with intestinal dendritic cells (DCs). It has been demonstrated that goblet cells can sample material from the gut lumen (McDole et al. 2012). The material is internalized by the goblet cells is then presented to closely allied immune tolerogenic DCs in the lamina propria (McDole et al. 2012).
The Epithelium: Enteroendocrine Cells
Enteroendocrine cells are less common in the colon than in the small intestine (Rothenberg et al. 2012). These cells produce and release a wide variety of peptides that modulate motility, intestinal cell secretion and absorption, glucose homeostasis, and a sense of satiety (Egerod et al. 2012; Sternini, Anselmi, and Rozengurt 2008). Although there was early speculation that they were derived from neural crest because of their shared features with neurons, enteroendocrine cells are derived from LGR5+ intestinal stem cells (Buczacki et al. 2013). Notch signaling plays an integral role in enteroendocrine cell differentiation. Notch signaling is inactivated in differentiated enteroendocrine cells, but these cells stimulate Notch signaling in neighboring cells to prevent their differentiation into enteroendocrine cells (May and Kaestner 2010; Noah and Shroyer 2013). In humans, chronic diarrhea and malabsorption have been associated with a paucity of enteroendocrine cells (Ohsie et al. 2009). These conditions include autoimmune polyglandular syndrome 1, enteroendocrine cell dysgenesis, and autoimmune enteropathy (Cortina et al. 2007; Du et al. 2012; Ohsie et al. 2009; Ruemmele et al. 2008; Wang et al. 2006).
In the colon, there are two distinct lineages of enteroendocrine cells: L-cells and enterochromaffin cells. Enteroendocrine L-cells express peptide YY (PYY) and glucagon-like peptide 1 (GLP-1) and may also co-express cholecystokinin and neurotensin (Sternini, Anselmi, and Rozengurt 2008). The apical surface of L-cells reach the lumen (termed open cells). Enteroendocrine cell secretion is activated by neuronal and endocrine signals as well as through “taste” or chemoreceptor cells for luminal contents. In the case of L-cells, glucose stimulates the release of GLP-1, which has a role in insulin regulation and glucose metabolism (Sternini, Anselmi, and Rozengurt 2008). Several GLP-1 agonists have shown efficacy, are marketed for the treatment of type II diabetes, and have not been associated with colonic toxicity (Bhat et al. 2013; Buse et al. 2013).
Enterochromaffin cells secrete serotonin (5-HT) with or without SP and do not generate the peptides expressed in L-cells. The apical membrane of enterochromaffin cells is not exposed to the lumen, and so these cells are termed closed cells and their products function in a paracrine or endocrine fashion (Sternini, Anselmi, and Rozengurt 2008). 5-HT of neuronal origin is important for motility but 5-HT derived from enterochromaffin cells is not. The role of 5-HT from enterochomaffin cells is diverse. Locally, 5-HT has been demonstrated to be proinflammatory, through interactions with DC (Ahern 2011; Li et al. 2011). In mouse studies, 5-HT derived from intestine has been demonstrated to be upregulated in fasting and results in increased lipolysis and increased gluconeogenesis in the liver (Sumara et al. 2012). It also seems to reduce blood glucose levels in mouse models of type II diabetes (Sumara et al. 2012). In addition, 5-HT has been shown to be important in bone remodeling and reducing bone formation (Cui et al. 2011; Yadav et al. 2008, 2009). Serotonin-reuptake inhibitors have been associated with osteoporosis in humans, which is in part a result of increased circulating gut-derived serotonin (Rizzoli et al. 2012; Wu et al. 2012).
Colonocytes and Xenobiotics
Several cancers have been demonstrated to have increased Notch activity through gain-of-function mutations or increased copy number (Schott et al. 2013). Efforts have been focused on blocking Notch signaling as a potentially useful method of treating cancers with these gain-of-function mutations (Schott et al. 2013; van Es et al. 2005). Notch signaling blockade could reduce cell proliferation and increase differentiation, potentially thwarting tumor growth and metastasis (Capaccione and Pine 2013; Schott et al. 2013; Shao, Huang, and Liu 2012; van Es et al. 2005). However, in mice with disruptions in intestinal Notch signaling and in both nonclinical and clinical trials of Notch inhibitors such as the gamma secretase inhibitors, goblet cell hypertrophy and hyperplasia have been observed (Figure 2; Capaccione and Pine 2013; Guilmeau et al. 2008; Milano et al. 2004; Wong et al. 2004). Similarly, dual inhibition of Notch 1 and Notch 2 using monoclonal antibodies causes goblet cell hypertrophy and hyperplasia in mice. However, recent studies suggest that the specific inhibition of Notch 2 (but not Notch 1) can reduce the goblet cell side effects (Wu et al. 2010). Goblet cell hyperplasia with intestinal Notch inhibition has also been associated with intestinal inflammation in mouse models, and chronic inactivation of Notch may result in colon cancer (Figure 2; Guilmeau et al. 2008). In sum, disruptions in mucus generation, glycosylation, and secretion may have a significant impact on the health of the colon (McGuckin et al. 2011; Molloy, Bouladoux, and Belkaid 2012).
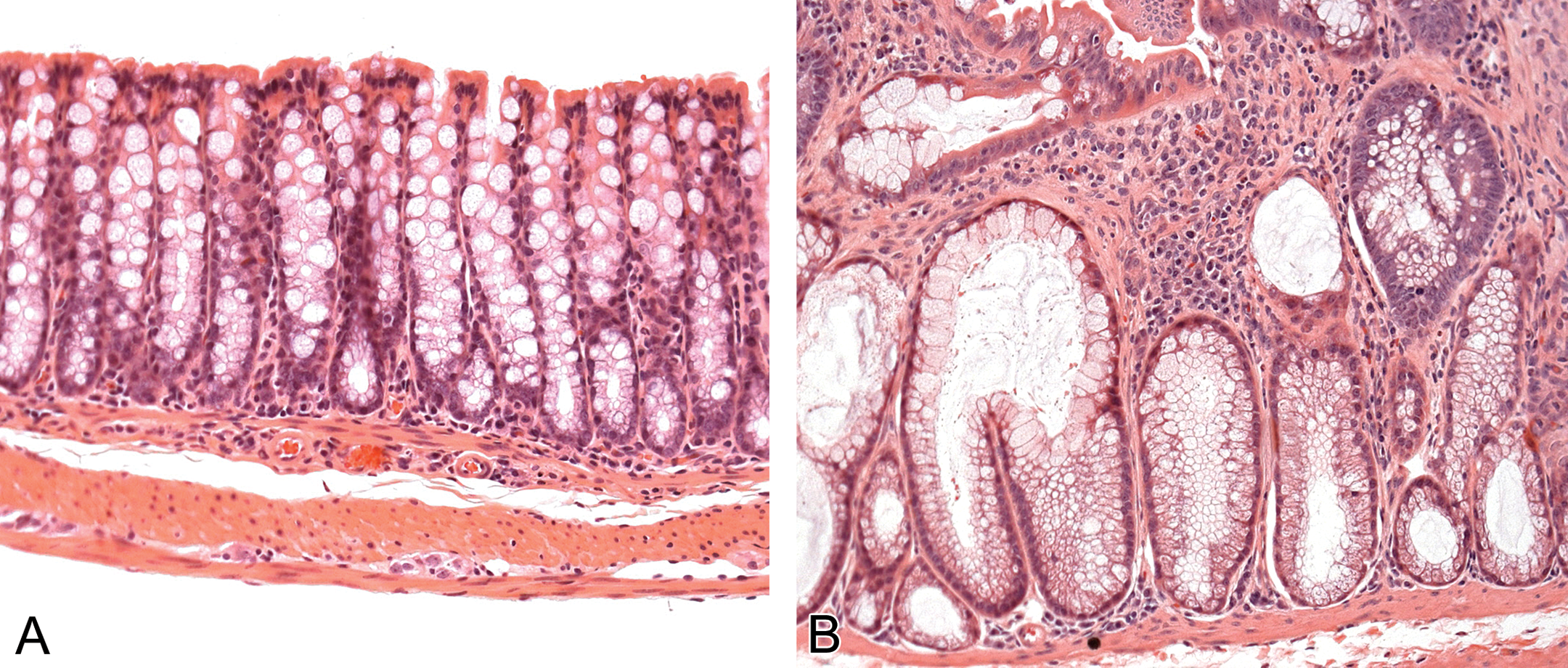
Photomicrographs depicting the mucosal morphology in the normal mouse middle colon (A) and a Pofut1 mutant mouse (mutant has inhibited Notch signaling in the intestine; B), both stained with H&E and imaged at 200× magnification. Note the goblet cell hypertrophy and hyperplasia, the foci of dysplasia, and the inflammatory infiltrates into the lamina propria.
Drugs used in cancer therapy routinely affect rapidly dividing cells and thus may have the unwanted effect of damaging normal proliferating cells, such as those of the intestinal crypt (Anilkumar et al. 1992; Moore 1986; Phillips and Sternberg 1975). The majority of these effects are present in the small intestine as the rate of proliferation of the cell population is lower in the colon than in the small intestine (Phillips and Sternberg 1975).
Colonic Immunity
The ability of the intestinal immune system to prevent microbial invasion while minimizing immune responses to commensal organisms is essential to maintaining colonic health (Hooper and Macpherson 2010; Kinnebrew and Pamer 2012; Wells et al. 2011). The colonic immune system, while in a state of active readiness, has unique adaptations to limit both microbial exposure and immune responses, ensuring a limited response to commensal organisms under normal conditions (Hooper and Macpherson 2010; Kinnebrew and Pamer 2012; Wells et al. 2011). The intestinal immune system is in large part independent of the systemic immune system. Immune responses in the intestine are driven by local DC, macrophages, T cells, and B cells, which are “educated” in the intestine or mesenteric lymph node. It is likely that many of the immune cells in the colon derive from cells “educated” in the small intestine (as opposed to in the colon, thymus, or spleen; Hooper and Macpherson 2010; Swiatczak and Rescigno 2012; Tanoue and Honda 2012).
The innate and the acquired immune systems function in a coordinated manner in the colon to create a functional colonic immune system, and DCs and macrophages often serve as the gatekeepers between the innate and adaptive immune system. The colonic immune system is essentially made up of three components (Hooper and Macpherson 2010). The first component is the mucous layer and its antimicrobial factors that limit microbial contact with IECs. The second is a rapid innate immune response to bacteria that breach the intestinal epithelium, which is initially mediated by macrophages. The third is an intestine-specific acquired immune response (Hooper and Macpherson 2010). Signaling by the intestinal epithelium to the cells of the lamina propria is essential to maintaining immunoregulatory function in the colon (Wells et al. 2011). Activation of the IECs by commensal organisms tends to drive immunotolerance in the intestine, whereas activation of cells in the lamina propria may drive a more proinflammatory response. Inappropriate innate and or adaptive immune responses can lead to inflammation and the entry of microbes into the systemic circulation (Kinnebrew and Pamer 2012).
Innate Responses
Organisms that breach the epithelium are generally phagocytosed and eliminated by macrophages in the lamina propria. While macrophages rapidly kill most bacteria entering the lamina propria, they do not activate a strong proinflammatory response to microbes, unlike in other tissues (Smith et al. 2011; Smythies et al. 2010). Stromal-derived TGF-β also prevents macrophages from secreting proinflammatory cytokines (Smith et al. 2011; Smythies et al. 2005). This muted inflammatory response is an adaptation to the intestinal microbiome and is essential to prevent chronic and damaging immune responses in the intestine (Cario 2013; Hooper and Macpherson 2010).
In the intestine as elsewhere, innate immune responses to microbes are mediated by pattern recognition receptors (PRRs), which are expressed on the IECs, phagocytic cells, endothelial cells, and fibroblasts. PRRs recognize foreign (nonself) conserved microbial structures termed pathogen-associated molecular patterns and damaged cells by damage-associated molecular patterns. PRRs fall into four families (1) toll-like receptors (TLRs); (2) C-type lectin receptors; (3) retinoic acid-inducible gene-I-like receptors; and (4) nucleotide-binding oligomerization domain (NOD)-like receptors (Jeannin, Jaillon, and Delneste 2008; Takeuchi and Akira 2010). In the colon, the best studied PRRs are the TLRs and NOD receptors (Abreu 2010). Depending on the combination of PRRs activated and the cell type (IECs vs. cells of lamina propria) and site at which they are activated (e.g., apical vs. basolateral membrane or intracellular), PRRs can drive the production of antimicrobial factors, induce or prevent inflammatory responses, and drive adaptive immune responses (Molloy, Bouladoux, and Belkaid 2012). Each microbe is sensed differently depending on the combination of PRRs it engages. The activation of PRRs in the IECs can have contradictory effects to those that occur in the underlying immune cells in the lamina propria (Abreu 2010). When the normal homeostatic immune mechanisms are not in place or are overwhelmed, the response to invading bacteria is marked by tissue destruction and inflammation.
Adaptive Responses
The development of the adaptive immune system in the GI tract is dependent on the microbiome (Chung et al. 2012). The formation of lymphoid follicles and the unique set of lymphocytes that are present in the intestinal mucosa are either not represented or underrepresented in gnotobiotic mice (El Aidy et al. 2012; Lee and Mazmanian 2010; Yamamoto et al. 2012). Intestinal DCs perform their function locally or within the mesenteric lymph node but not at immune sites outside of these areas (Mann et al. 2013; Rescigno 2010). In the small and large intestines, there are a number of types of DCs that can be identified by their expression of various markers (Mann et al. 2013; Matteoli et al. 2010; Rescigno 2010). DCs work in collaboration with the IECs and macrophages to modulate the intestinal adaptive immune system and control the balance between anti-inflammatory intestinal regulatory T cell (iTreg—CD4+FOXP3+) and proinflammatory effector T cell (e.g., Th1, Th17, and CD8+ T cells) differentiation.
In the healthy colon, tolerance to microbes is the dominant immune response (Hooper and Macpherson 2010). The central mechanism by which the adaptive immune system maintains host–microbial homeostasis is through the suppression of immune responses against commensal microbes (immune tolerance; Cario 2013; Hooper and Macpherson 2010). The intestine has a unique set of T regulatory cells, termed iTregs, which are different from the classic Tregs (nTreg) that originate in the thymus. iTregs are intestine-derived and intestinal antigen-specific T suppressor cells. Differentiation of naive CD4+ cells to iTregs can be through direct interactions with DC or through DC and stromal-secreted factors such as TGF-β, interleukin (IL)-10, IL-2, prostaglandin E2, and retinoic acid (Rescigno 2010; Smith et al. 2011; Smythies et al. 2005, 2010; Tanoue and Honda 2012; Worthington et al. 2011). DCs that are CD103+ and express indoleamine 2, 3 dioxygenase drive tolerogenic responses by promoting the production of iTregs and downregulating Th17 cell differentiation (Matteoli et al. 2010; Rescigno 2010). IEC release of IL-10, TGF-β, and thymic stromal lymphopoietin (TSLP) also drives tolerogenic responses by DCs (Duerr and Hornef 2012; Roda et al. 2010). TSLP is constitutively expressed by IEC, but its expression appears to be induced by commensal organisms (Mann et al. 2013). DCs can also downregulate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent gene expression (typically proinflammatory) in other cells (Smythies et al. 2005, 2010; Worthington et al. 2011). Certain species of bacteria, including Clostridium strains and Bacteroides fragilis, have been demonstrated to increase the number of Treg cells in the colon, which is consistent with data suggesting beneficial effects of probiotics (Ivanov and Littman 2010; Nutsch and Hsieh 2012; Round and Mazmanian 2010). Disruptions in immune tolerance can result in abnormal responses to commensal bacteria, and this mechanism is suspected to be a component in the development of IBD (Manichanh et al. 2012; Mann et al. 2013).
The differentiation of naive T cells into proinflammatory Th1 or Th17 helper T cells is dependent primarily on the cytokines released by antigen presenting cells (APCs) that respond to pathogens via PRRs and the types of DCs activated. For example, CX3CR1+CD70+ DCs in the small intestine tend to promote inflammation by promoting Th17 differentiation (Bain et al. 2013). DCs also play an important role in producing proinflammatory cytokines such as IL-6, IL-12, and IL-18 and can drive differentiation into Th1-type T cells (Mann et al. 2013). In particular, ongoing inflammation in the gut appears to promote proinflammatory DC cell formation and cell recruitment to the intestine (Mann et al. 2013).
A clear understanding as to how the intestinal immune system identifies when and when not to respond to microbes is likely related to several factors, including the type and combination of PRRs expressed and engaged on specific cell types (Mann et al. 2013), the location of PRR engagement (e.g., at the IEC apical or basolateral surface), the number and type of microbes encountered, and/or the mechanisms of microbial exposure (e.g., through ulcerations; Wells et al. 2011). One factor that may be important in maintaining a tolerogenic phenotype in the intestine is mucin. As previously stated, abnormal mucin production or glycosylation can result in intestinal inflammation (Heazlewood et al. 2008; Kim and Ho 2010; Moehle et al. 2006; Nishida et al. 2012; Resta-Lenert et al. 2011; Van der Sluis et al. 2006; Velcich et al. 2002; Yang et al. 2008). A recent study has produced intriguing data that could help explain the mechanism by which disrupted and abnormal production or glycosylation of mucins can result in intestinal inflammation. Using both in vitro and in vivo experiments, studies from the lab of A. Cerutti (A. Cerutti, personal communication, 2013) demonstrated that MUC2 is important in promoting a tolerogenic response by intestinal DCs and IECs. Their data show that under normal conditions, MUC2 colocalizes with bacteria and lipopolysaccharide (LPS) in intestinal DC. The LPS/bacteria–MUC2 complex in DCs promotes a tolerogenic response by DCs through the downregulation of proinflammatory cytokines such as IL-12 and upregulation of anti-inflammatory cytokines such as IL-10. MUC2 alone also appears to promote the expression of IL-10 by IECs and DCs. In the absence of MUC2, the intestinal DCs and IECs have decreased expression of IL-10 and increased expression of proinflammatory cytokines such as IL-12 (A. Cerutti, personal communication, 2013). These data are provocative and may have a significant impact on our understanding of chronic inflammatory diseases of the intestine and how to treat them.
Immunologic Dysfunction and Xenobiotics
Compounds that modulate the immune system have the potential to significantly disrupt colonic homeostasis. Immune modulators to treat diseases have become increasingly common. These modulators, which include small molecules and biologics, treat IBD and autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, or function as cancer chemotherapeutics. Several of the immune modulating biologics on the market are associated with colonic adverse effects ranging from diarrhea and colitis to ischemic colitis (Salk et al. 2013a, 2013b). In the case of immunomodulators that block immune tolerance, termed immune checkpoint inhibitors, “immune-related adverse events” have been reported (Corsello et al. 2013). These inhibitors function by blocking immune tolerance to tumor antigens and have demonstrated success in treating certain forms of cancer. Examples include blockers of the cytotoxic T-lymphocyte antigen 4 receptor (Ipilimumab) and programmed death 1 (Tremelimumab, BMS-936558), both of which have been associated with colitis and/or diarrhea (Corsello et al. 2013). The proposed mechanism for the colitis is a loss of local tolerance that favors a more proinflammatory environment in the colon.
MAPK inhibitors are also associated with chronic colitis in nonhuman primates in nonclinical studies (author’s personal experience). Dogs are uniquely sensitive to MAPK inhibitors and develop lymphocyte necrosis and apoptosis in the gut-associated lymphoid tissue followed by colonic and cecal mucosal necrosis and hemorrhage (Morris et al. 2010). The mechanism of this toxicity is at least in part due to the greater dependence of canine B lymphocytes on the p38α-signaling cascade for proliferation and survival as compared to other species (Morris et al. 2010). A recent study in mice also demonstrated that pharmacologic inhibition of MEK-ERK-signaling enhances Th17 cell differentiation, thus promoting a proinflammatory environment (Tan and Lam 2010).
Prostaglandin inhibitors (e.g., nonsteroidal anti-inflammatory drugs/NSAIDs) have well-documented effects on blood flow in the small intestine and stomach, resulting in intestinal ulceration and bleeding (Geboes, Hertogh, and Ectors 2006). However, NSAIDs can also lead to collagenous or lymphocytic colitis (which resembles IBD; Kaufmann and Taubin 1987), exacerbate preexisting IBD, or complicate diverticular disease (i.e., perforation or bleeding; Gibson, Whitacre, and Ricotti 1992; Huber, Ruchti, and Halter 1991; Kaufmann and Taubin 1987). Several other drugs including ranitidine, ticlopidine, lansoprazole, flutamide, bentazepam, tardyferon, and vinburnine have also been suggested to have caused lymphocytic/collagenous colitis (Cappell 2004).
Vasculature
The colonic vasculature derives from both the superior and the inferior mesenteric arteries. The small intestine receives approximately 2.5-fold more of the cardiac output than does the colon (De Fontgalland et al. 2008). Blood flow to the intestine increases during digestion (Matheson, Wilson, and Garrison 2000), and this increase in blood flow after ingestion of food can be 2-fold over baseline and continue for 2 to 3 hr (Matheson, Wilson, and Garrison 2000). The increased blood flow is primarily directed through the mucosal microvasculature to aid in the absorption of nutrients and the removal of waste. Blood flow in the intestine, as elsewhere, is controlled by a variety of neural factors, hormones, and paracrine, autocrine, and metabolic factors (e.g., pH, PCO2, PO2; Matheson, Wilson, and Garrison 2000). Neuronal control of vascular tone allows for rapid changes in blood flow in response to changing conditions in the intestine during digestion. Enteric nerves and sympathetic nerve fibers connect to the intestinal microvasculature; however, it appears that sympathetic innervation dominates the tone of intestinal vessels (De Fontgalland et al. 2008). Vasodilatory agents include somatostatin, neuropeptide Y, SP, and VIP as well as histamine and NO (De Fontgalland et al. 2008; Matheson, Wilson, and Garrison 2000).
The intestinal vasculature also functions in the targeting of immune cells into the gut; Intestinal endothelial cells have specific mechanisms to promote the localization of immune cells to the intestine. Adhesion molecules (e.g., mucosal vascular addressin cell adhesion molecule 1 and α4β7) are important in lymphocyte migration into the mucosa of the large and small intestines, as are a variety of chemokines and their receptors (e.g., CXCL12-CXCR4; De Fontgalland et al. 2008).
Disrupted Blood Flow and Xenobiotics
A number of xenobiotics have been associated with GI vascular events (Casella et al. 2009; Geboes, Hertogh, and Ectors 2006; Park, Gunn, and Harrison 2012; Salk et al. 2013a, 2013b; Zeino, Sisson, and Bjarnason 2010). Ischemic colitis has been documented, though rarely, in patients treated with TNF-α inhibitors and type I interferons for hepatitis C and multiple sclerosis (Okanoue et al. 1996; Salk et al. 2013a, 2013b; Tada et al. 1996). There is also a report of colonic ischemia after an accidental overdose of verapamil (Perbet et al. 2009). The mechanism by which these drugs cause ischemic colitis is uncertain, but vasospasm or local hypertension is most likely (Salk et al. 2013a, 2013b). In fact, any drug that causes severe mesenteric vasoconstriction, such as pseudoephedrine or vasopressin, has the potential to result in intestinal ischemia (Dowd et al. 1999; Klestov, Kubler, and Meulet 2001; Lambert, de Peyer, and Muller 1982). Drug-associated vascular thrombosis such as with oral contraceptives and bevacizumab may also be a cause for colon ischemia (Gurbuz et al. 1994; Schutz et al. 2011). Local thrombosis may also be caused by drugs given orally or intrarectally, such as with Kayexalate that is used to treat hyperkalemia (Harel et al. 2013)
Conclusions
Colonic homeostasis is based on complex interactions between the mucosal epithelial cells, immune cells, vasculature, stromal cells, nervous system, and microbes. Disruptions in any component of the colonic function and microenvironment may have substantial local and systemic consequences. Direct and indirect effects of pharmaceuticals may result in adverse findings in laboratory animals and humans.
Footnotes
Acknowledgments
The authors wish to thank Dr. Anna Velcich and Dr. Leonard Augenlicht for the use of materials for photomicrographs.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.