
Abstract
A panel of pathologists (Panel) was formed to evaluate the pathogenesis and human relevance of tumors that developed in the fundic region of rat stomachs in carcinogenicity and mechanistic studies with alachlor and butachlor. The Panel evaluated stomach sections stained with hematoxylin and eosin, neuron-specific enolase, and chromogranin A to determine the presence and relative proportion of enterochromaffin-like (ECL) cells in the tumors and concluded all tumors were derived from ECL cells. Biochemical and pathological data demonstrated the tumor formation involved a nongenotoxic threshold mode of action (MOA) initially characterized by profound atrophy of the glandular fundic mucosa that affected gastric glands, but not surface epithelium. This resulted in a substantial loss of parietal cells and a compensatory mucosal cell proliferation. The loss of parietal cells caused a marked increase in gastric pH (hypochlorhydria), leading to sustained and profound hypergastrinemia. The mucosal atrophy, together with the increased gastrin, stimulated cell growth in one or more ECL cell populations, resulting in neoplasia. ECL cell autocrine and paracrine effects led to dedifferentiation of ECL cell tumors. The Panel concluded the tumors develop via a threshold-dependent nongenotoxic MOA, under conditions not relevant to humans.
Keywords
Introduction
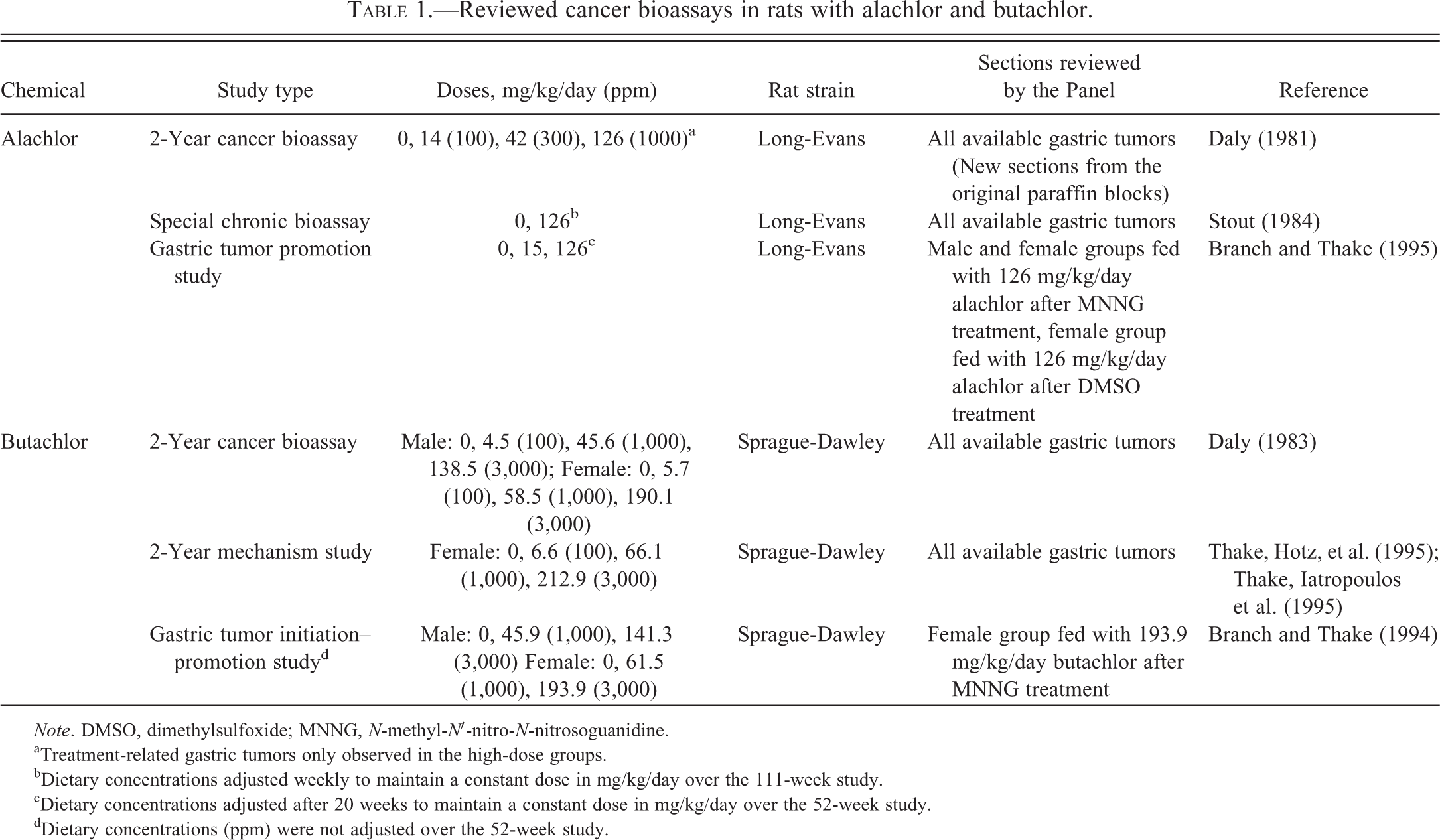
Alachlor and butachlor are chloroacetanilide herbicides that share substantial structural and functional similarities. In long-term carcinogenicity studies (Table 1), at doses that met or exceeded the maximum tolerated dose, rats developed gastric tumors that presented a diagnostic dilemma (Hard 2003). Although a similar threshold response was noted in both sexes, at comparable doses female rats developed more tumors than male rats.
Reviewed cancer bioassays in rats with alachlor and butachlor.
Note. DMSO, dimethylsulfoxide; MNNG, N-methyl-N′-nitro-N-nitrosoguanidine.
aTreatment-related gastric tumors only observed in the high-dose groups.
bDietary concentrations adjusted weekly to maintain a constant dose in mg/kg/day over the 111-week study.
cDietary concentrations adjusted after 20 weeks to maintain a constant dose in mg/kg/day over the 52-week study.
dDietary concentrations (ppm) were not adjusted over the 52-week study.
Since there was substantial morphologic variability within and among the gastric tumors as diagnosed in the original carcinogenicity studies (Daly 1981, 1983), reevaluations of the gastric tumors from the original bioassays for both alachlor and butachlor were previously conducted in an attempt to identify the tumor type involved and to determine whether the morphologic characteristics of tumors from the two original studies with alachlor and butachlor were comparable (Hard and Iatropoulos 1994; Hard et al. 1995; Heydens et al. 1999; International Agency for Research on Cancer [IARC] 2003). Also, a series of histochemical and immunohistochemical stains were used to identify features that would aid in identification of the cell/cells of origin. Those evaluations demonstrated that morphologic characteristics of the gastric tumors were consistent for both chemicals (Hard and Iatropoulos 1994; Hard et al. 1995; Heydens et al. 1999) and further revealed that the features present in gastric tumors from both chemicals were consistent with the wide variety of histological appearances previously described in enterochromaffin-like (ECL) cell tumors in rodents (Chen and Hakanson 2003; Fossmark et al. 2004; Lewin and Appelman 1996; Martinsen et al. 2003; Waldum, Brenna, and Sandvik 1998; Waldum, Peterson, and Brenna 1992). However, several different laboratories and pathologists were involved in the evaluation and diagnosis of tumors from those studies. As a result, diagnoses varied.
These inconsistencies have resulted in questions related to the current state of the science regarding the interpretation and significance of the original findings. In order to provide a consistent, consensus opinion, a panel of pathologists (Panel) was recently convened to review all gastric tumors from all rat cancer studies conducted with alachlor or butachlor. Consensus diagnoses were established for all gastric tumors using standard criteria. Additionally, studies performed to elucidate the mode of action (MOA) of gastric tumor formation (Table 1) were summarized and reevaluated according to defined criteria (Table 2), and literature related to ECL cell tumors was assessed. The consensus diagnoses were that all tumors, except some of those initiated with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), were derived from ECL cells. Furthermore, the consensus opinion of the Panel was that the MOA was not relevant for assessing human risk.
Morphologic, phenotypic, and immunohistochemical classifications for ECL cell hyperplasia and ECL cell tumors.
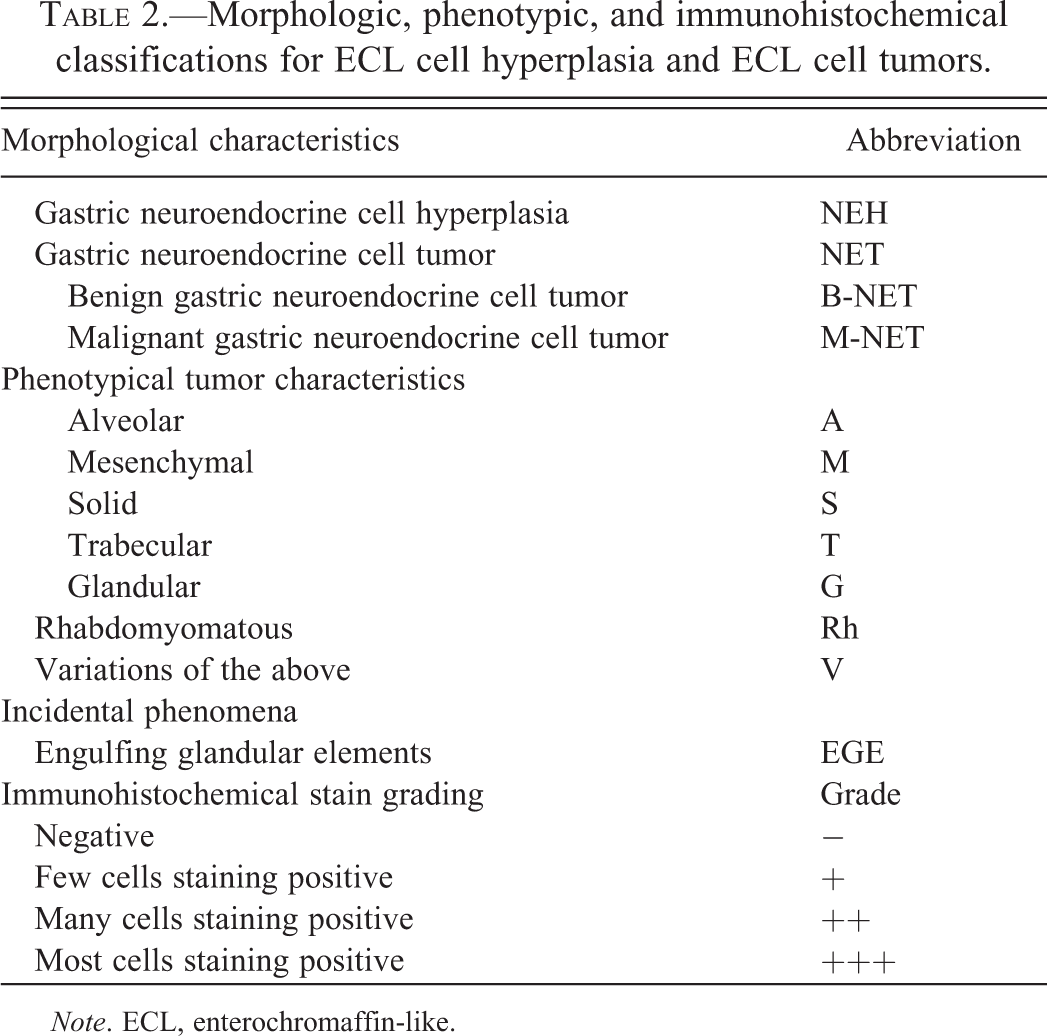
Note. ECL, enterochromaffin-like.
Method
The following are the alachlor and butachlor studies in which gastric neoplasms were induced and subsequently reevaluated by the Panel.
Two-year Cancer Bioassays with Alachlor and Butachlor
Long-term dietary administration of alachlor and butachlor to Long-Evans (LE) and Sprague-Dawley (SD) rats, respectively, in the original carcinogenicity studies (Table 1) resulted in treatment-related gastric tumors only in the high-dose groups (Daly 1981, 1983). Based on numerous pathological and biochemical changes, the top doses in those original studies met or exceeded the maximally tolerated dose. Treatment-related gastric tumors were present only in the fundic region of the stomach and occurred more commonly in female than in male rats.
Special Chronic Bioassay/2-year Mechanism Studies with Alachlor and Butachlor
A long-term (22 month) bioassay was also conducted (2-year mechanism study, Table 1) by feeding butachlor to female rats to help elucidate the MOA that was responsible for induction of the gastric tumors in the original cancer study (Thake, Hotz, et al. 1995; Thake, Iatropoulos et al. 1995). Histopathologic, histochemical, and immunohistochemical evaluations were performed using (primarily fundic) stomach tissue from rats; additional end points measured included mucosal thickness, mucosal cell proliferation, gastric acid secretion, serum gastrin levels, and receptor binding (for cholecystokinin/gastrin binding sites in tumors).
Similarly, a mechanism study (special chronic bioassay, Table 1) with alachlor was also conducted (Stout 1984). Although the primary purpose of the study was to further characterize ocular lesions observed in the original bioassay, the study was also modified to further characterize the stomach tumors noted in the original study. Gastric tumor incidence in animals treated with alachlor until spontaneous death or sacrifice at the end of the study was 4% (3/70) for males and 61% (19/31) for females. Originally, the stomach tumors observed in the alachlor special chronic bioassay and the butachlor mechanism study were organized into six different diagnostic categories, but upon reevaluation the tumors were all categorized as poorly differentiated gastric carcinoids (Tables 3 and 4) and the morphologic diversity of the observed tumors was noted as being consistent with phenotypes of previously reported tumors that originated from ECL cells.
Alachlor: Special chronic bioassay.
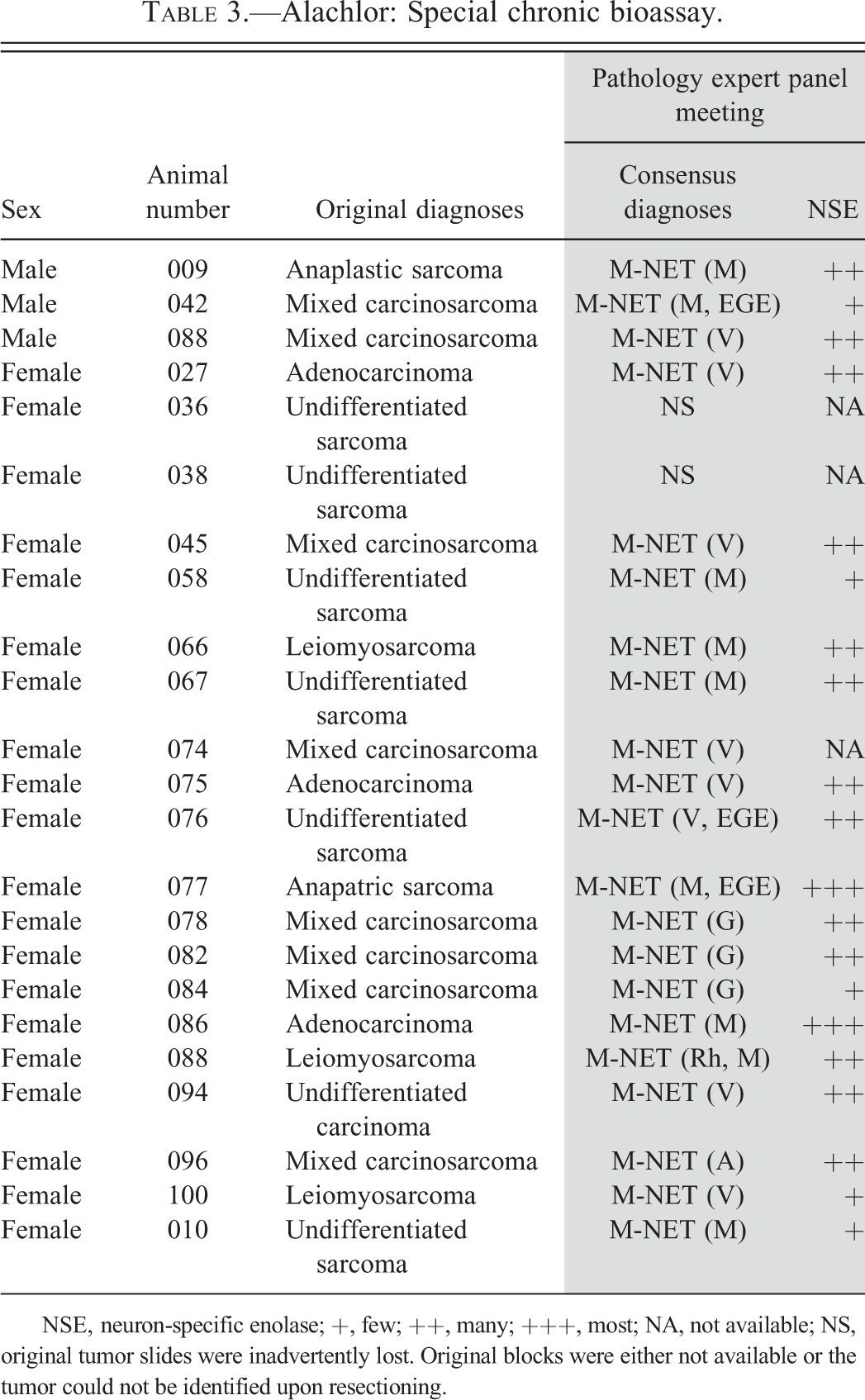
NSE, neuron-specific enolase; +, few; ++, many; +++, most; NA, not available; NS, original tumor slides were inadvertently lost. Original blocks were either not available or the tumor could not be identified upon resectioning.
Butachlor: 2-year mechanism study.
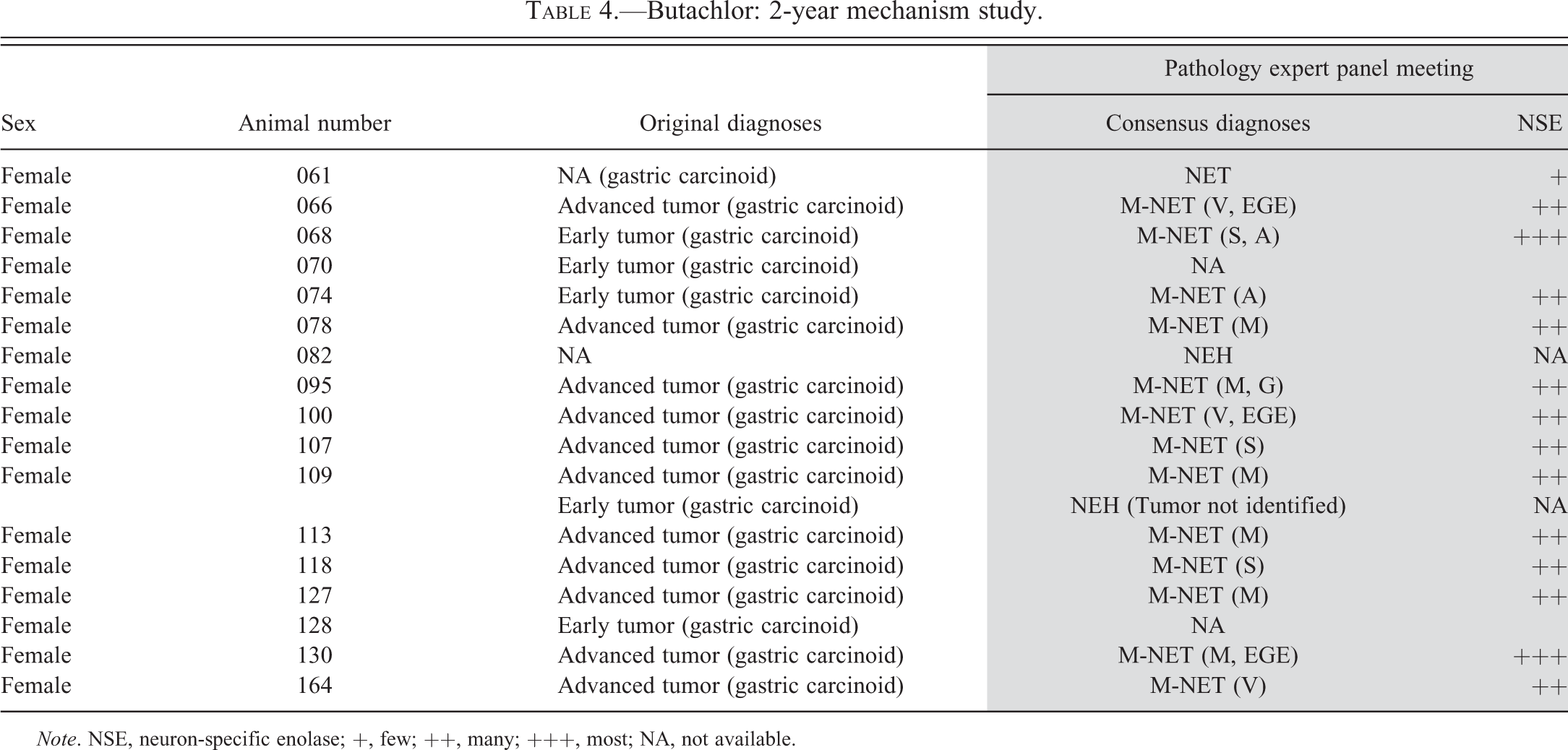
Note. NSE, neuron-specific enolase; +, few; ++, many; +++, most; NA, not available.
Gastric Tumor Initiation and/or Promotion Studies with Alachlor and Butachlor
In an effort to address the initiation and/or promotion potential of alachlor and butachlor, an initiation–promotion study was conducted with butachlor, using MNNG as the standard gastric genotoxic initiating agent. In addition, a promotion study was performed with alachlor (Table 1; Branch and Thake 1994, 1995). The results of the studies demonstrated that butachlor did not have initiating activity and that both alachlor and butachlor had promotional activity which was predominantly restricted to the fundic region of the stomach. As such, the conclusions from those reports support other data indicating that the tumors form via a nongenotoxic MOA subject to a threshold in a less acidic gastric environment.
Reevaluation of Alachlor and Butachlor Gastric Neoplasms
Sections of alachlor- and butachlor-associated gastric tumors were reviewed by the Panel (Drs. Furukawa, Harada, Iatropoulos, and Thake). The original slides containing gastric tumors from the original 2-year cancer bioassay with alachlor (Daly 1981) could not be located by the contract laboratory archivist. Therefore, new sections from the original paraffin blocks were prepared and those slides were reviewed by the Panel.
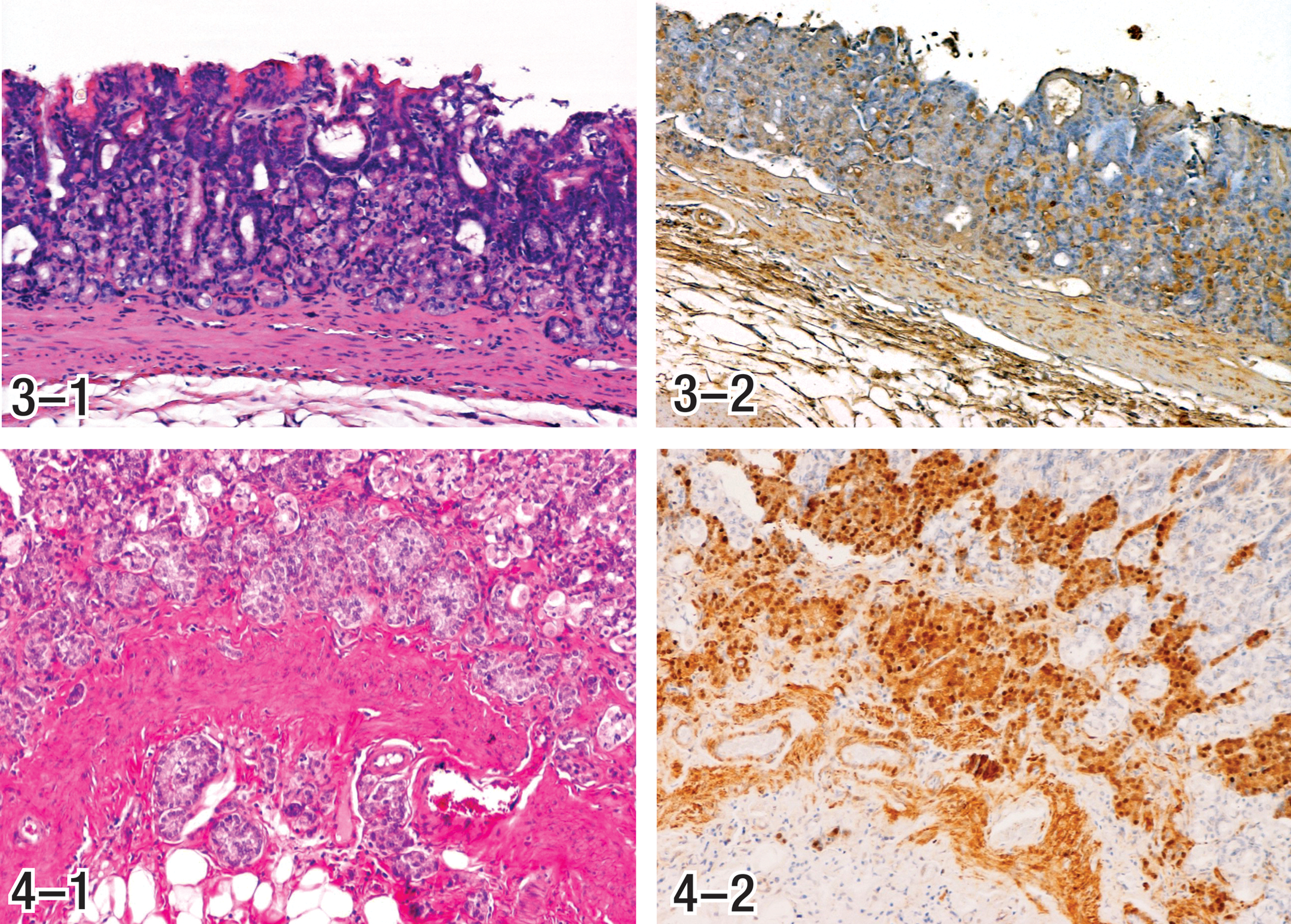
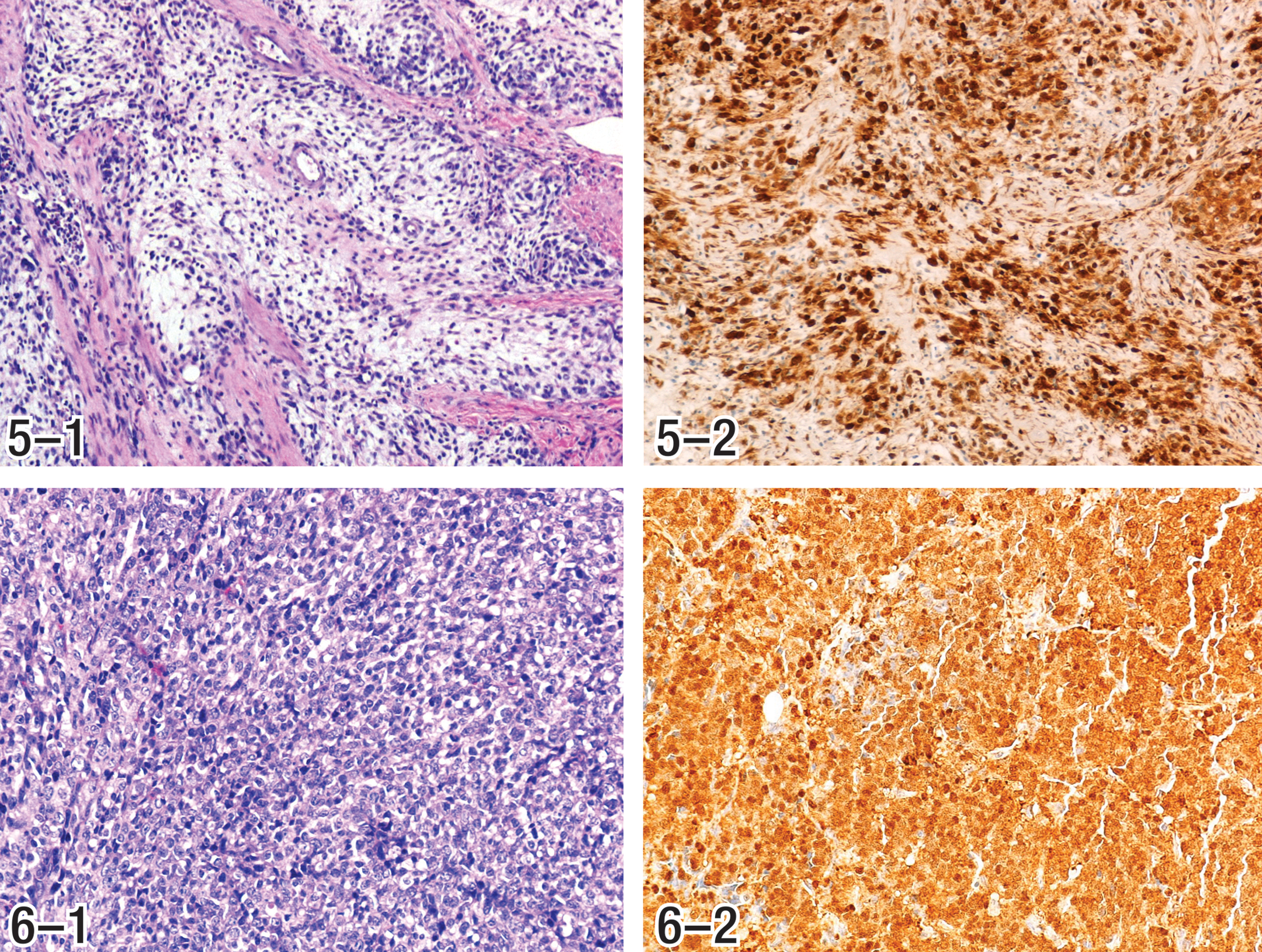
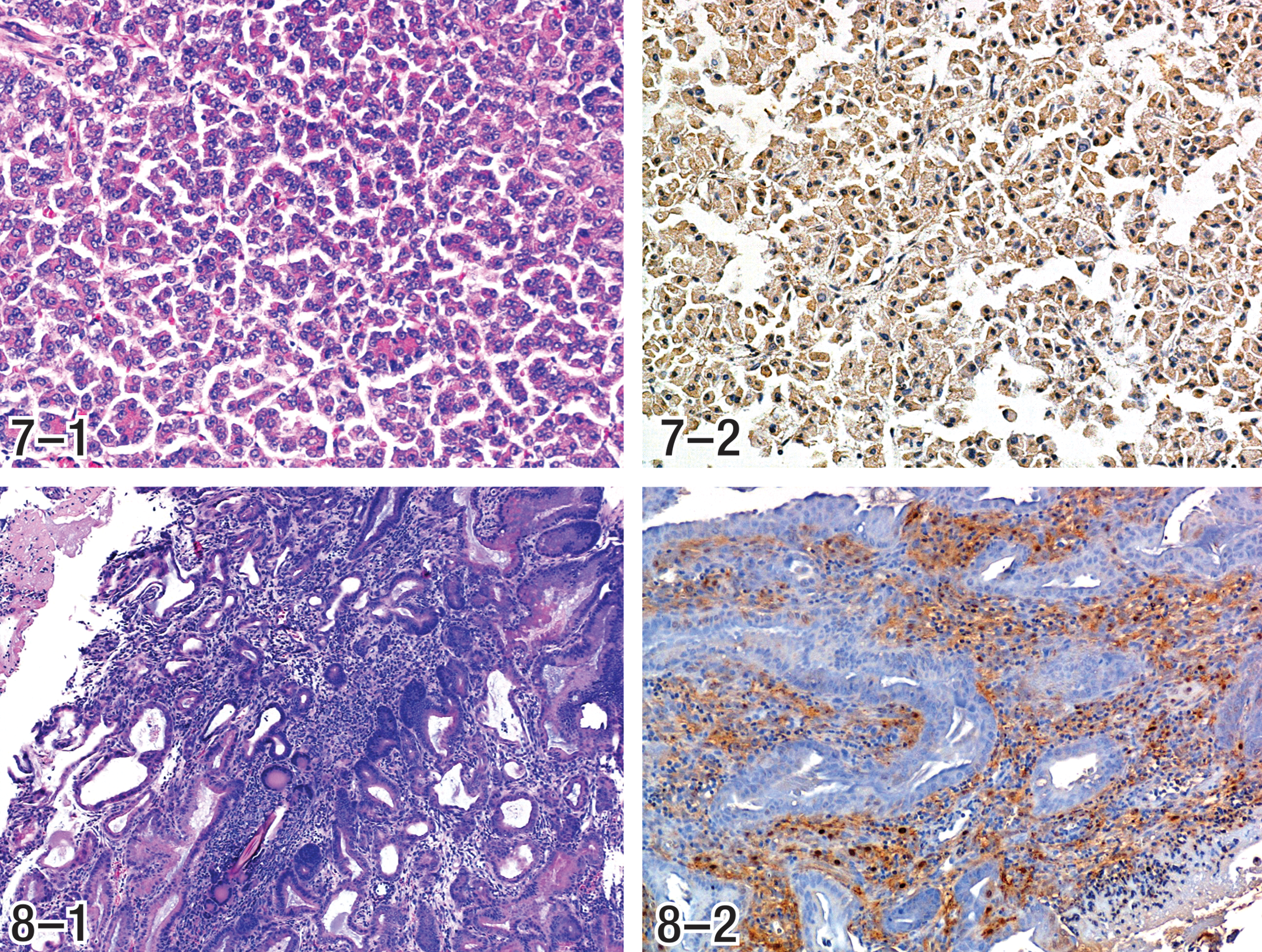
The Panel evaluated the available tumors and established standardized diagnoses based on specific criteria for ECL cell hyperplasia and neoplasia, as summarized in Table 2. Example photomicrographs depicting each of the phenotypic characteristics of ECL cell tumors, identified in Table 2 and described in the following section, are presented in Figures 1 through 10. Consensus diagnoses were established for each tumor. Tumor classifications were based on cell of origin as determined by staining with neuron-specific enolase and chromogranin A (CGA) and/or major morphologic characteristics (Table 2). Immunohistochemical stains were available for most but not all hyperplasias and tumors. However, for tumors without immunohistochemically stained sections, the Panel members were able to develop consensus opinions by bridging of results, based upon common phenotypic histopathologic characteristics observed in hematoxylin and eosin (H&E) stained sections.
Results
Diagnoses in Long-term Studies (2-year Cancer Bioassays, Special Chronic Bioassays, and 2-year Mechanism Study)
Although tumors that resulted from exposure to both alachlor and butachlor displayed various phenotypes, using the criteria in Table 2, all alachlor- and butachlor-induced gastric tumors and hyperplasias that occurred in long-term carcinogenesis studies were concluded to be of ECL cell origin. The results of this reevaluation are presented in Tables 3 through 6. Because infiltrative growth was observed in the majority of these tumors, the majority of the tumors were diagnosed as malignant neuroendocrine cell tumors. Phenotypes represented in these tumors are summarized in Table 9. The tumors observed in the alachlor feeding study tend to have a higher percentage of the glandular phenotype, as well as a higher percentage of tumors associated with more engulfing glandular elements (EGE). However, the majority of the phenotypic patterns of gastric tumors resulting from exposure to alachlor and butachlor were mesenchymal or variations and were similar to one another.
Alachlor: 2-year cancer bioassay.
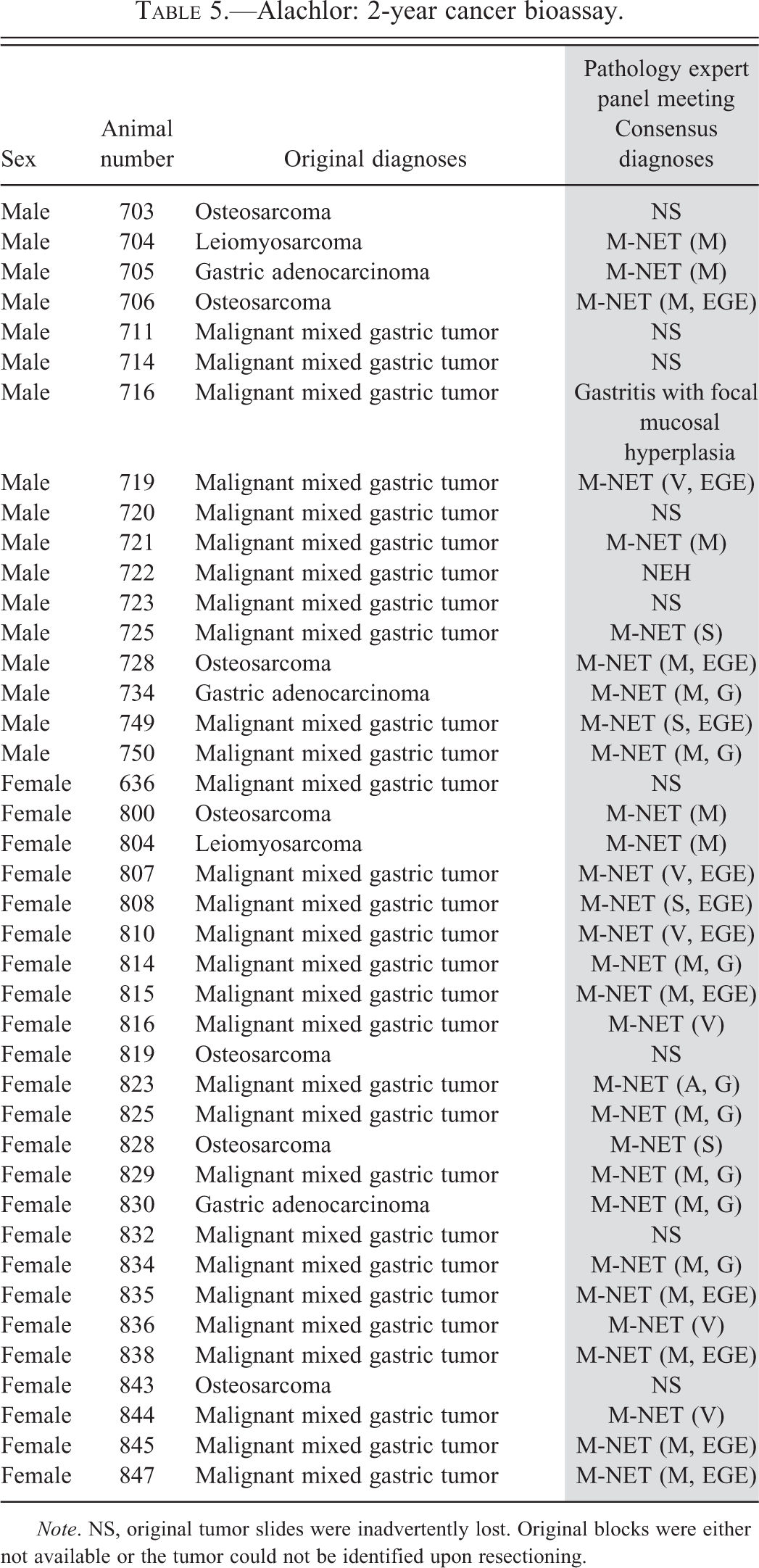
Note. NS, original tumor slides were inadvertently lost. Original blocks were either not available or the tumor could not be identified upon resectioning.
Butachlor: 2-year cancer bioassay.
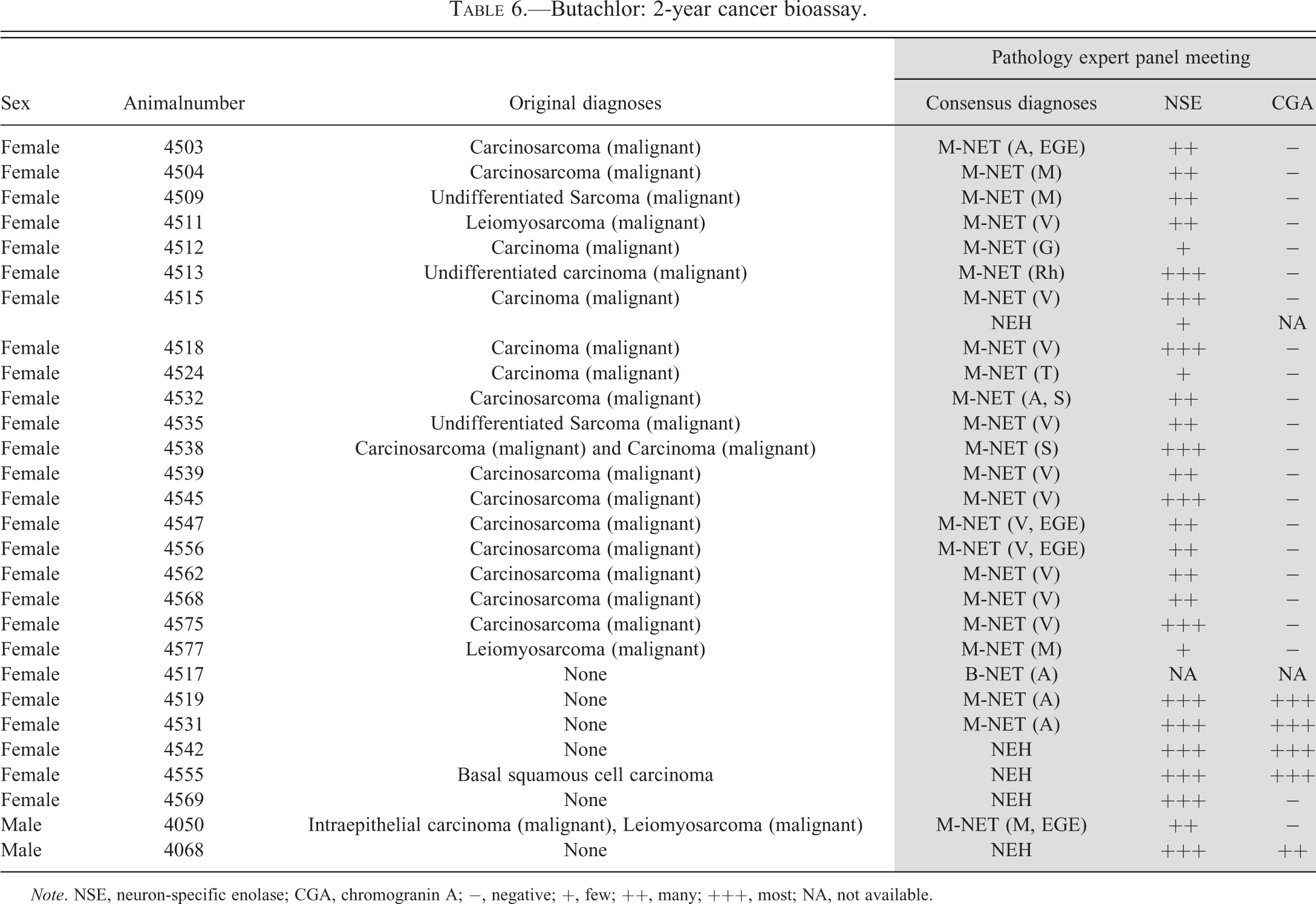
Note. NSE, neuron-specific enolase; CGA, chromogranin A; −, negative; +, few; ++, many; +++, most; NA, not available.
In the 2-year cancer bioassay with alachlor, 9 of the 41 original alachlor-induced gastric tumors could not be identified in the newly prepared slides (Table 5). However, the original diagnosis for the tumors from the missing blocks and slides were identical to the original diagnoses for other tumors examined by the Panel and reclassified as ECL cell tumors. This strongly suggests that the missing tumors were similar to those examined by the Panel and, as such, were likely to also have been ECL cell tumors.
Diagnoses in Gastric Initiation–Promotion/Promotion Studies
The gastric tumor initiation–promotion/promotion
Alachlor: Gastric tumor promotion study.
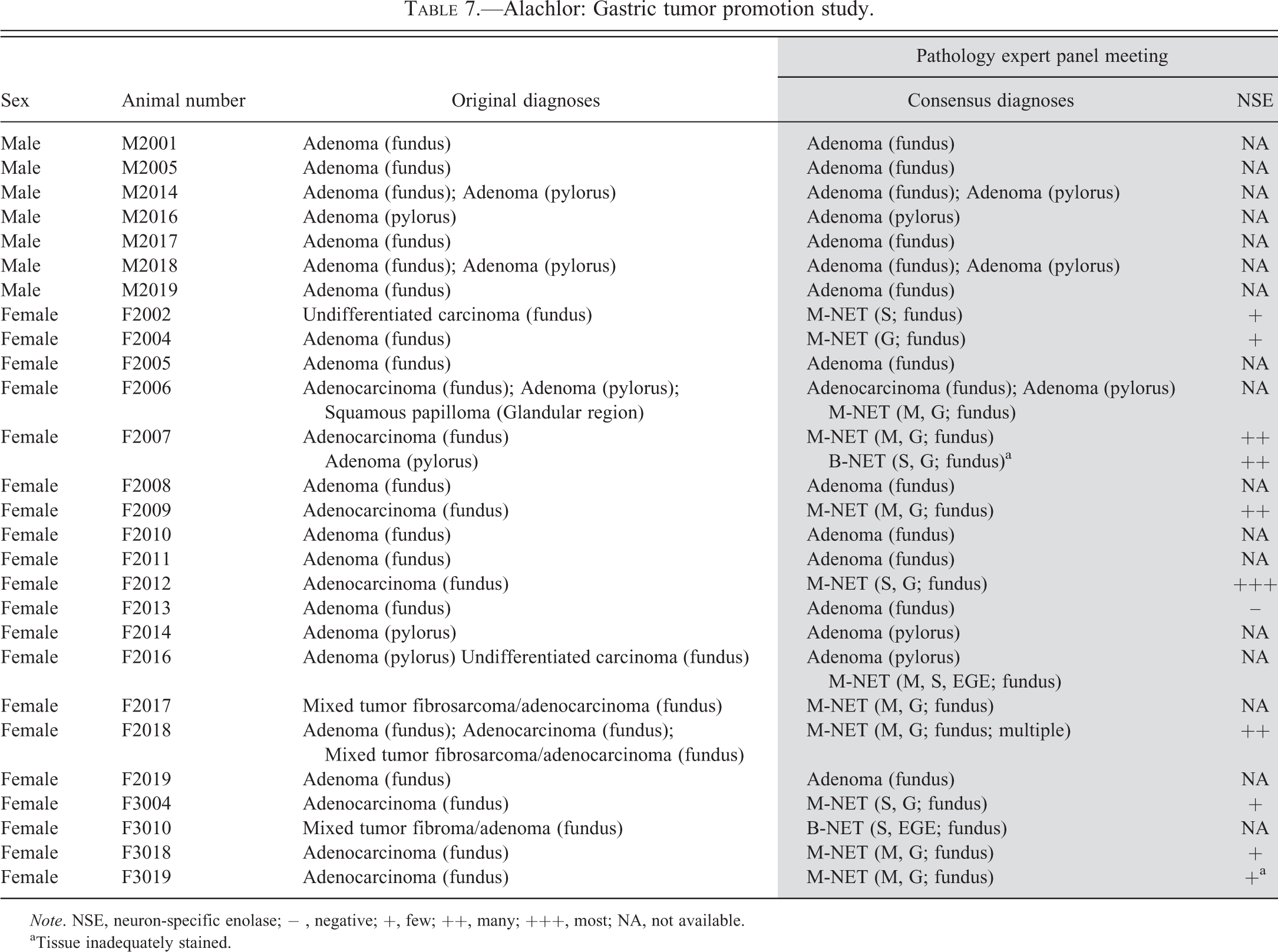
Note. NSE, neuron-specific enolase; − , negative; +, few ; ++, many; +++, most; NA, not available.
aTissue inadequately stained.
Butachlor: Gastric tumor initiation–promotion study.
Summary of panel pathological diagnoses.
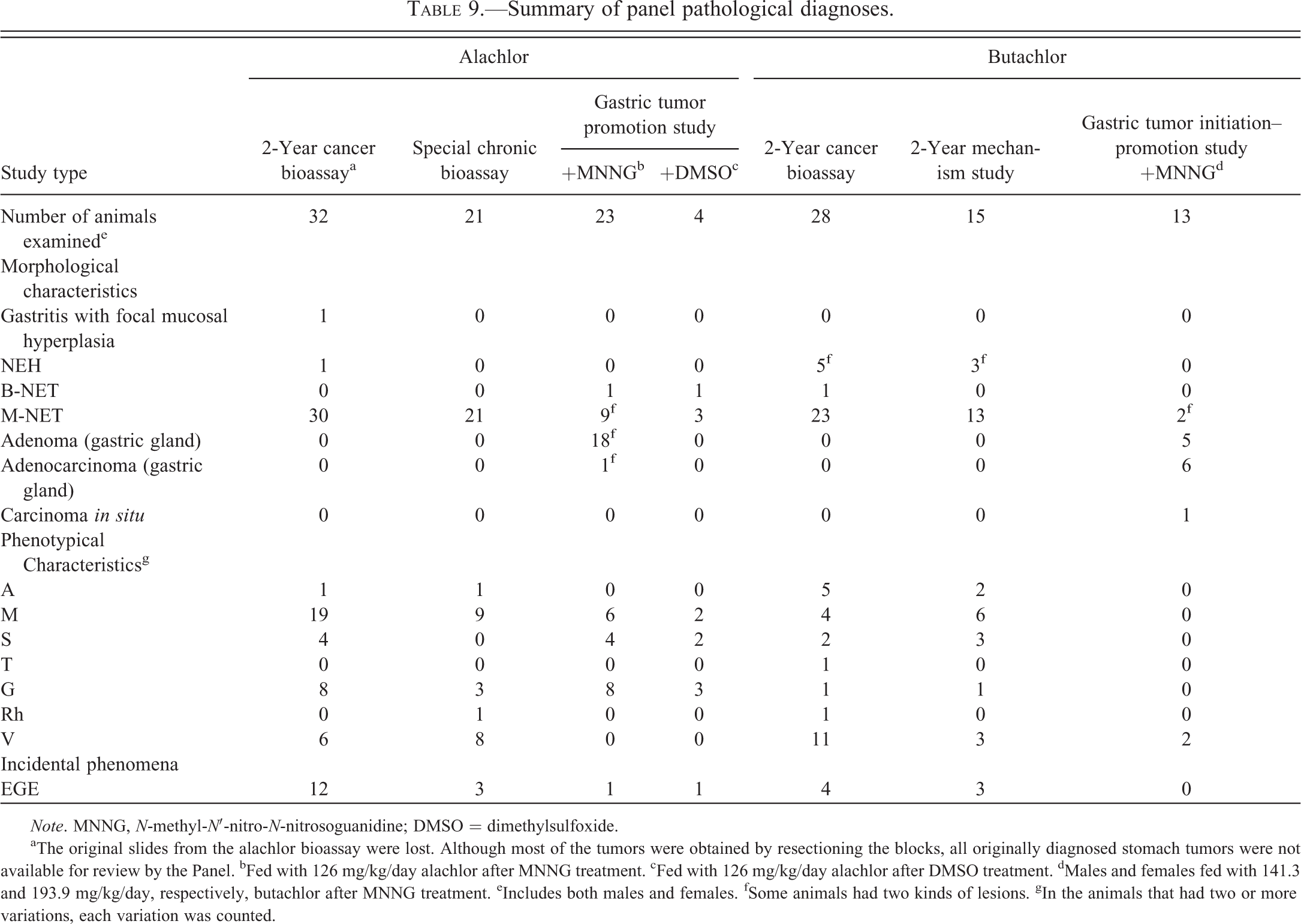
Note. MNNG, N-methyl-N′-nitro-N-nitrosoguanidine; DMSO = dimethylsulfoxide.
aThe original slides from the alachlor bioassay were lost. Although most of the tumors were obtained by resectioning the blocks, all originally diagnosed stomach tumors were not available for review by the Panel. bFed with 126 mg/kg/day alachlor after MNNG treatment. cFed with 126 mg/kg/day alachlor after DMSO treatment. dMales and females fed with 141.3 and 193.9 mg/kg/day, respectively, butachlor after MNNG treatment. eIncludes both males and females. fSome animals had two kinds of lesions. gIn the animals that had two or more variations, each variation was counted.
Fundic mucosal atrophy was a prominent feature in these studies, as it was in the previous long-term studies with these substances. Features of the neuroendocrine cell tumors induced by MNNG plus butachlor or MNNG plus alachlor were generally similar to those induced by alachlor or butachlor alone.
Tumors that developed in the fundic region of the stomach in the initiation–promotion study with butachlor, and the promotion study with alachlor, were diagnosed as being of either ECL cell or epithelial cell origin. NSE stains were available for some, but not all, rats in the alachlor promotion study. Where they were available, the morphologic characteristics and positive NSE stain demonstrated the ECL cell origin of the tumors. On the other hand, as would be expected from initiation with the genotoxic carcinogen MNNG, adenomas and adenocarcinomas were additionally observed in the studies. If the NSE stain was not available and the primary feature of the tumor was adenoma- or adenocarcinoma-like, those diagnoses were retained. It is noteworthy that 4 fundic tumors were present in animals given alachlor (126 mg/kg/day) only for 1 year. All 4 tumors were of ECL cell origin based on positive NSE staining. Butachlor (3,000 ppm; 141.3 mg/kg/day for males, 193.9 mg/kg/day for females) alone, without MNNG initiation, did not result in gastric tumors in the initiation–promotion study.
Discussion
Based upon a review of all available treatment-related gastric tumors induced in long-term studies and gastric tumor initiation–promotion/promotion studies performed with alachlor and butachlor, the Panel concluded that all gastric tumors, except some of those initiated with MNNG, were derived from ECL cells at various stages of differentiation (Table 9). The majority of the phenotypic patterns expressed in the alachlor- and butachlor-induced gastric tumors were mesenchymal or variations and were similar to one another. The Panel developed a consensus MOA of ECL cell tumorigenesis by alachlor and butachlor from the proliferative, biochemical, and neoplastic data reviewed from all alachlor and butachlor chronic rat studies.
MOA of ECL Tumorigenesis in Alachlor- and Butachlor-treated Rats
Nine key events (Table 10) are believed to be involved in the development of alachlor- and butachlor-induced ECL cell tumors with variable phenotypic characteristics. Substantial mechanistic work has been performed to verify the importance of each of these key steps, as discussed below.
Key events of ECL cell tumorigenesis in alachlor and butachlor studies.

Note. ECL, enterochromaffin-like.
1. Profound mucosal atrophy in the fundic region
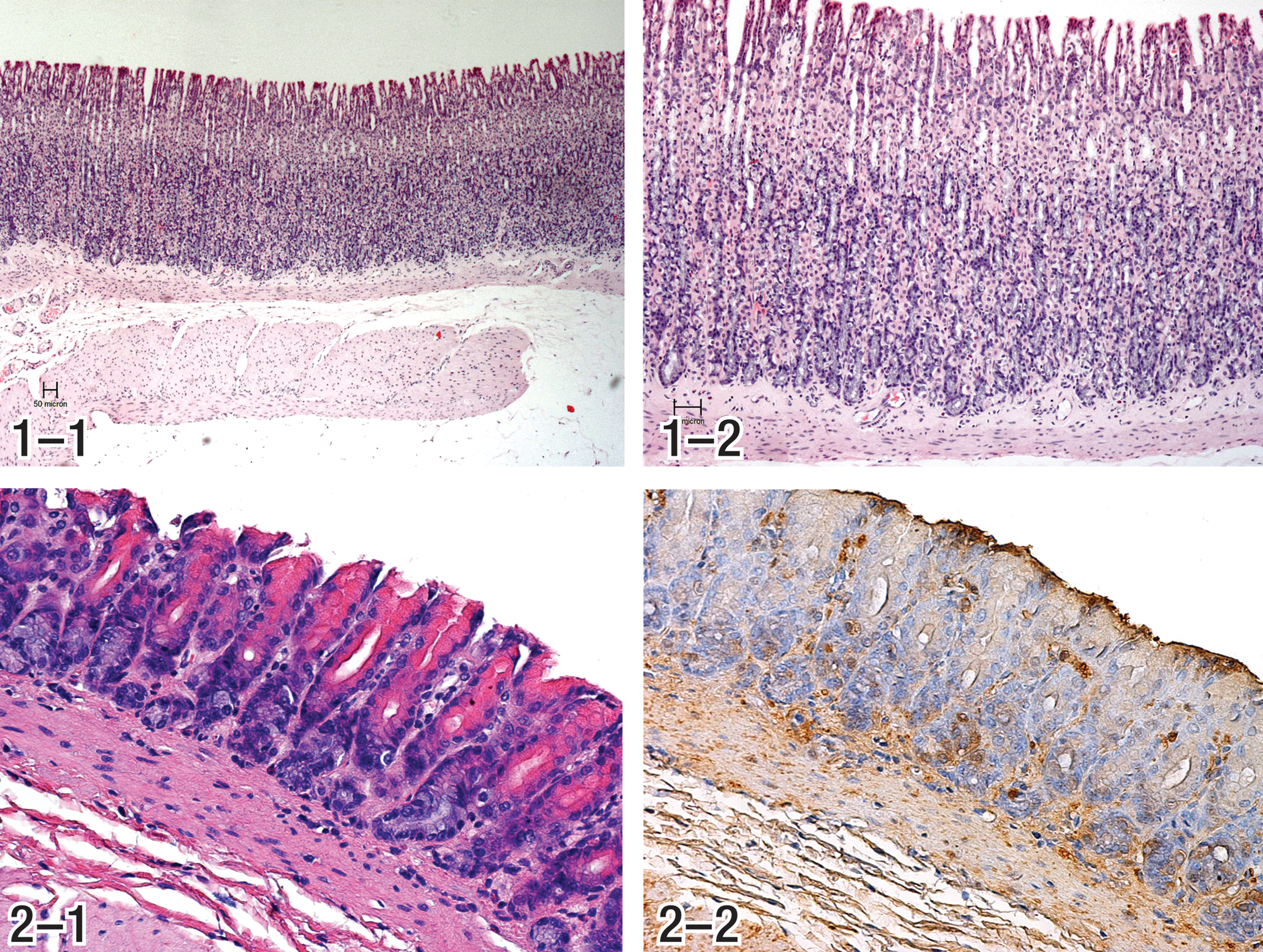
The first step toward alachlor and butachlor gastric neoplasia is represented by the development of a profound mucosal atrophy in the fundic region of the stomach, as seen in rats given 3,000 ppm (212.9 mg/kg/day) butachlor for 20 months, where a profound mucosal atrophy with substantial loss of glandular epithelium in the fundic region was evident while the surface epithelium was generally intact (Figures 1a, b, and 2a, b). Mucosal thickness measurements in the butachlor mechanism study demonstrated that decreases in mucosal thickness became evident beginning at 30 days and continued through 20 months, at which time the mucosal thickness of treated (3,000 ppm) rats averaged less than half that of controls (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Mucosal thickness measurements in rats from a previous 26-month study also demonstrated decreased mucosal thickness in females given 3,000 ppm (190.1 mg/kg/day) and in those given 1,000 ppm (58.5 mg/kg/day), although in the latter animals the decrease was substantially less than for the 3,000 ppm group (control = 0.75 mm, mid-dose = 0.59 mm, high dose = 0.35 mm; Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Mucosal atrophy was confined to the fundic mucosa with no apparent involvement of the pyloric mucosa (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). The much milder mucosal atrophy in the mid-dose rats (1,000 ppm) was not sufficient to result in hypoacidity and hypergastrinemia, as discussed below. Similar to that observed in the cancer bioassays, there was also profound mucosal atrophy in the fundic region of butachlor and alachlor high-dose treated groups (Table 1) from the gastric tumor initiation–promotion and promotion studies, respectively (Branch and Thake 1994, 1995). It is highly unlikely that humans could be continuously exposed to the large doses of alachlor and butachlor necessary to induce sustained profound mucosal atrophy in the stomach.
Results from all carcinogenicity studies with alachlor and butachlor demonstrate that profound fundic mucosal atrophy is the initial precancerous change necessary (but not sufficient) for the formation of ECL cell tumors operating via compensatory ECL cell hyperplasia. At a dose level that induced mild mucosal atrophy, but was not sufficient to induce hypoacidity, there was no hypergastrinemia and there were no gastric tumors (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995).
2. Loss of parietal cells
In rats, the parietal cells comprise approximately 40% of the cells in the oxyntic gland (Chen and Hakanson 2003; Iatropoulos 1986). As such, the profound mucosal atrophy in the glandular stomach of rats in high-dose groups treated with alachlor and butachlor resulted in substantial loss of parietal cells, which are responsible for acid secretion in the glandular stomach. The parietal cell depletion was especially prominent, although all elements of the deeper mucosa also appeared to be affected (Figures 2-1 and 2-2). In rats bearing gastric tumors, the deeper mucosa in some nontumor areas was comprised of epithelium that appeared to be poorly differentiated, such that the normal elements of the deeper fundic mucosa (i.e., parietal, chief, or ECL cells) were not recognizable (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Parietal cell number measurements in the alachlor gastric tumor promotion study demonstrated that after 1 year of treatment with 126 mg/kg/day, the numbers of parietal cells were profoundly reduced to 7% of the control value (Branch and Thake 1995; Heydens et al. 1999; Tatematsu and Thake 1996).
3. Hypochlorhydria
Mechanistic studies in female rats treated for approximately 21 months with butachlor demonstrated that the loss of parietal cells in the fundic mucosa resulted in hypochlorhydria/hypoacidity. Mean pH of stomach content had increased to 5.69 in the 3,000-ppm group at 21 months of age while that of rats from the 100- and 1,000-ppm (6.6 and 66.1 mg/kg/day, respectively) groups were at control level (i.e., approximately pH 2.7). The increased pH correlated with the degree of mucosal atrophy and with the substantial decrease in gastric acid secretion (approximately 78% decrease in secretion) in rats from the 3,000-ppm dose group. A much smaller decrease (approximately 37%) in gastric acid secretion was noted in rats from the 1,000-ppm dose group (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995).
Similarly, in the promotion study with alachlor, after 1 year of treatment of rats receiving 126 mg/kg/day, the females had approximately a 50% increase in stomach pH (2.87–4.50), while the males experienced a less dramatic increase in stomach pH. Much greater influence on gastric acid secretion was noted in the alachlor study, with a 7-fold and 4-fold decrease in HCl secretion in high-dose males and females, respectively (Branch and Thake 1995).
4. Hypergastrinemia
In response to the loss of parietal cells and accompanying hypoacidity, G cells in the pyloric region of the stomach were stimulated to secrete gastrin through a paracrine feedback mechanism (Figure 11). Gastrin acts on ECL cells to stimulate cell division and histamine secretion. The histamine secreted by ECL cells stimulates parietal cells to increase acid production (Chen and Hakanson 2003), which results in feedback control of gastrin secretion by G cells. If sufficient numbers of parietal cells are lost, increased gastrin production fails to regulate the hypoacidity and hypergastrinemia ensues.
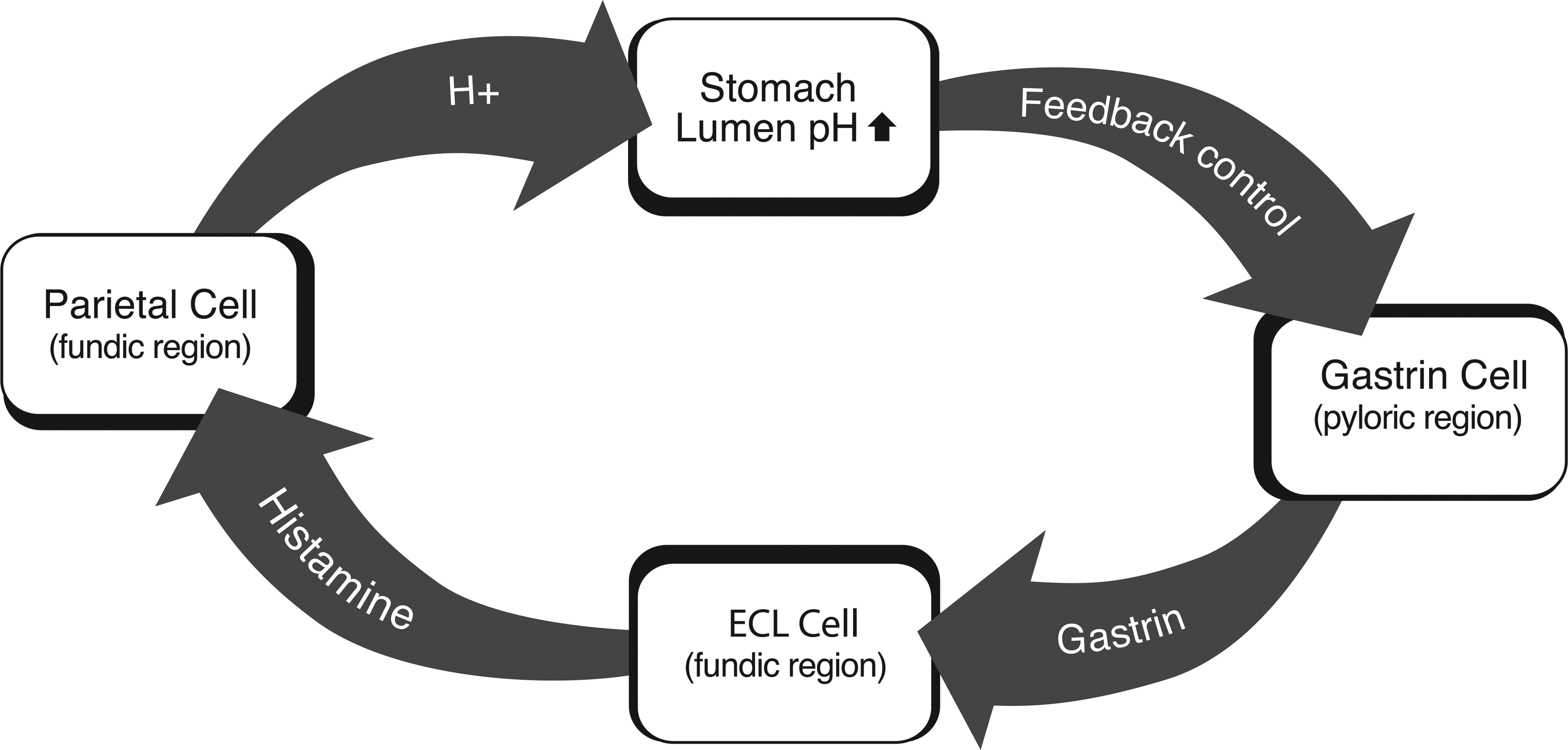
Hypergastrinemia has been associated with gastric ECL cell tumors in rats. There have been numerous studies of ECL cell tumor induction as a result of treatment with potent inhibitors of gastric acid secretion (Chen and Hakanson 2003; Fossmark et al. 2004; Havu 1986; Hirth et al. 1988; Poynter et al. 1985). In the butachlor mechanism study, it is clear that hypoacidity is necessary, but not sufficient, for the induction of the gastric tumors. Although mucosal atrophy was present to a mild extent in animals given 1,000-ppm butachlor, the mucosal changes were not adequate to induce gastric hypoacidity (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). However, the more severe mucosal atrophy that was present in rats given 3,000-ppm butachlor resulted in substantial gastric hypoacidity (Thake, Hotz, et al. 1995; Thake, Iatropoulos et al. 1995). The absence of hypoacidity in the 1,000-ppm dose group resulted in normal gastrin levels, whereas the hypoacidity that occurred in the 3,000-ppm group resulted in greatly increased (8.5× control values; 75 pg/ml in control vs. 642 pg/ml in the high-dose group) serum gastrin values (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Therefore, although butachlor induced a mild mucosal atrophy in the 1,000-ppm dose group, there was no hypoacidity or hypergastrinemia (serum gastrin in mid-dose was 98 pg/ml vs. 75 pg/ml in control), so there were no resulting tumors. These significant and critical data demonstrate the role of gastrin in the alachlor and butachlor induction of gastric ECL cell tumors.
Results from the mechanism study with butachlor are corroborated by measurements of serum gastrin levels in high-dose rats from the alachlor promotion study, where serum gastrin levels were elevated to approximately 700% and 1,700% of control values in males and females, respectively (Branch and Thake 1995; Heydens et al. 1999).
5. Sustained cell proliferation in the gastric fundic mucosa
Gastrin is a potent cell growth factor for the gastric fundic mucosa (Rozengurt and Walsh 2001). It has been shown that gastrin has a direct trophic action on ECL cells and on cells in the proliferative zone of the gastric mucosa (Brenna et al. 1995). Substantially increased cell proliferation has been demonstrated in gastric mucosa of butachlor-treated rats at the tumorigenic dose level (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Cell proliferation in the mucosa was markedly increased apparently to compensate for loss of cells in the glandular epithelium (Figures 3-1 and 3-2). Elevated levels of gastrin observed in alachlor and butachlor studies would be expected to be trophic to ECL cells in which there is an increased level of proliferation, regardless of their stage of maturity.
Approximately 7-fold to 9-fold increases in proliferation (as measured by proliferating cell nuclear antigen analysis) were observed at the base of the fundic glands after treatment with butachlor for 60 days and for 20 months, while no changes were observed in the pyloric mucosa. Changes in proliferation were much less when measured at the neck of the fundic mucosa (Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). However, this must be interpreted in light of the restructured mucosal architecture in a mucosa whose thickness has been substantially decreased. The proliferative zone that is normally at the mucosal neck would have collapsed toward the base, due to the loss of gastric gland epithelium. The increased proliferation at the base of the epithelium may also, at least in part, reflect a primary effect of gastrin on the adult (mature) ECL cell population.
6. ECL cell hyperplasia
ECL cell tumors that occurred in alachlor and butachlor studies were generally obtained at terminal sacrifice in the 2-year bioassays when the tumors were in the late stages of maturity. For that reason, ECL cell hyperplasia was not often observed as a component of these neoplasms or in the nontumor mucosa in those animals. However, more comprehensive examination of the gastric fundic mucosa from females in the highest (3,000 ppm) dose group from the butachlor bioassay and the butachlor mechanism study revealed 11 early lesions, 3 of which were determined to be areas of hyperplastic ECL cells. The remainders were determined to be very early ECL cell tumors; it was clear, however, that the early tumors were a continuation of development from hyperplasia (Hard et al. 1995). Hyperplasia is a constant feature of the gastric mucosa in cotton rats that develop ECL cell tumors spontaneously or following treatment with loxtidine (Fossmark et al. 2004; Martinsen et al. 2003) and is presumed to be a precursor of the neoplasia. This also appears to be true for the alachlor and butachlor gastric tumors and early lesions observed as described above are apparently the earliest prodromal manifestations of ECL cell neoplasia.
7. ECL cell neoplasia
During reevaluation of butachlor gastric tumors, it was determined that they were of ECL cell origin. In an attempt to identify the ECL cell differentiation stage/stages of tumor origin, early tumors observed in the butachlor mechanism study were evaluated. Because only a few early tumors were noted in the study, it was not possible to definitively identify at which point in the stage of ECL cell differentiation neoplastic transformation was initiated (ECL stem cells, undifferentiated ECL cells, or mature ECL cells). However, as noted in Table 9, the current review definitively identified all treat-ment-related gastric tumors observed after alachlor and butachlor treatment (except those initiated with MNNG) as being derived from ECL cells. Furthermore, the consensus of the Panel was that ECL cell tumors resulting from alachlor and butachlor treatment likely originate from ECL cells in different stages of differentiation.
8. ECL cell paracrine effects
Gastrin is well known for its role in stimulating acid secretion through its effect on ECL cell histamine production (Walsh 1990). It is a potent cell growth factor in the gastric epithelium and also may be a factor in maintenance of gastric mucosa and in neoplastic transformation (Rozengurt and Walsh 2001). Gastrin has also been shown to regulate other cell processes including apoptosis, cell migration, cell invasion, tissue remodeling, and angiogenesis (Burkitt, Varro, and Pritchard 2009). However, there is evidence that the trophic effect of gastrin on oxyntic mucosa may be due to paracrine effects, that is, effects that are mediated by substances from the ECL cell (Burkitt, Varro, and Pritchard 2009; Fossmark, Qvigstad, and Waldum 2008; Waldum, Brenna, and Sandvik 1998) that act locally on other mucosal elements. Substances such as basic fibroblast growth factor (bFGF) and histamine are also produced by ECL cells (Bordi et al. 1994; Waldum, Brenna, and Sandvik 1998), and both have an angiogenic effect (Fox, Gatter, and Harris 1996). The production of bFGF in tumors of ECL cell origin may be responsible for the desmoplastic proliferation (Fox, Gatter, and Harris 1996), and along with histamine may promote angioblastic proliferations (Fox, Gatter, and Harris 1996; Waldum, Brenna, and Sandvik 1998). This is consistent with the desmoplasia that is prominent in many alachlor and butachlor gastric neoplasms (Hard and Iatropoulos 1994), and with the angiogenesis that is observed in some of these neoplasms. Likewise, the production of certain matrix metalloproteinases is influenced by gastrin and may be responsible for tissue remodeling and invasion in humans and animals with ECL cell neoplasms (Burkitt, Varro, and Pritchard 2009; Wroblewski et al. 2002). Therefore, in addition to its direct trophic effect on fundic ECL cells, gastrin may play an indirect role in carcinogenesis of the fundic mucosa by inducing the release of paracrine factors from the ECL cells which may influence the responses of other mucosal cells.
9. Dedifferentiation of ECL cell tumors to fully developed tumors with variable phenotypic characteristics
ECL cell tumors have the capacity to dedifferentiate as they grow (Fossmark, Qvigstad, and Waldum 2008; Fossmark et al. 2005) and are known to display a wide variety of histological appearances (Lewin and Appelman 1996). This is particularly relevant since in cotton rats the ECL cell tumors can have an adenocarcinoma phenotype with neuroendocrine differentiation (Fossmark et al. 2004, 2005) and ECL cell tumors with similar characteristics can also be induced in cotton rats with the H2 blocker loxtidine (Fossmark et al. 2004). A glandular tumor phenotype was also described in CD rats after long-term administration of loxtidine (Poynter et al. 1985). The tumors are induced by hypergastrinemia resulting from hypoacidity and further progress through dedifferentiation. Cellular features that present similar to an adenocarcinoma phenotype are also present in many alachlor- and butachlor-induced gastric tumors and have historically contributed to the general confusion regarding their classification. It has been shown that gastric neoplasms in humans which result from severe atrophic gastritis and which have been previously classified as adenocarcinomas have been shown to have neuroendocrine differentiation and may be of ECL cell origin (Qvigstad et al. 2002; Waldum et al. 2008). Dedifferentiation in alachlor- and butachlor-induced gastric tumors also apparently explains the reason for the anaplastic appearance of tumor cells, the presence of adenomatous- or mesenchymal-like phenotypes, and loss of tumor staining by ECL cell markers as the alachlor and butachlor tumors progress.
Immunohistochemical staining has been used with variable results in alachlor- and butachlor-induced tumors. A large variety of histochemical or immunohistochemical staining has been undertaken; however, the most meaningful results were obtained from the NSE and CGA stains. The poorly differentiated cell type in many of these tumors was apparently largely responsible for the variability in histochemical and immunohistochemical staining. In those tumors for which there was little staining, scattered cells or nests of cells were generally positive which is the expected result with dedifferentiated cells (Hard et al. 1995; Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995). Immunohistochemical markers are often positive in only a portion of the cells in ECL cell tumors in cotton rats apparently due to the dedifferentiation that occurs (Martinsen et al. 2003). This is also true for classical neuroendocrine tumors in other organs (Creutzfeldt et al. 1973; Gould et al. 1983; Sumiyoshi et al. 1998) and has been attributed to gradual loss of features specific for the neuroendocrine cells due to dedifferentiation (Creutzfeldt et al. 1973; Gould et al. 1983). In those instances in alachlor- and butachlor-induced tumors where there were early lesions that had not yet progressed to the dedifferentiated phenotype, staining by NSE and CGA was generally prominent. Therefore, with alachlor- and butachlor-induced tumors for which the cell type specific features of ECL cells persist, the NSE stain is consistently positive. This stain becomes variable and less consistent as the tumors progress and dedifferentiation occurs, with loss of cytologic features responsible for staining by this technique. As such, although a variety of phenotypes are expressed in fully developed gastric tumors induced by alachlor and butachlor, all of the tumors originate from ECL cells at various stages of differentiation.
In summary, the initial injury leading to ECL cell tumors is gastric mucosal atrophy, while the direct induction of tumors is due to the resulting compensatory gastrin stimulation of ECL cells in the development of gastric tumors induced by alachlor and butachlor. The alteration of the neuroendocrine architecture of the fundic mucosa, although initially reversible, when subject to sustained chronicity, leads to cellular dysplasia of proliferating ECL cells at different stages of maturity and finally to cellular autonomy consistent with neoplasia (Fossmark, Qvigstad, and Waldum 2008; IARC 2003; Martinsen et al. 2003; Waldum et al. 2008). Phenotypic variability in the alachlor and butachlor tumors is due to dedifferentiation with redifferentiation and to ECL cell-induced paracrine effects.
Cellular Origins of ECL Cell Tumors Induced by Treatment with Alachlor and Butachlor
It has been shown that multipotential stem cells are present in the gastric mucosa of adult rodents (Bjerknes and Cheng 2002). Other work also suggests that committed progenitor cells for parietal, zymogen, and mucous cells retain proliferative capability in the gastric mucosa of rodents (Bordi et al. 1994). It is also known that rodent ECL cells retain the capacity for proliferation (Waldum, Brenna, and Sandvik 1998). Gastrin has been shown to target the proliferation of ECL cells at different stages of maturity, as well as other stem cells in the rat stomach (Ryberg et al. 1990). Moreover, it has been shown that a common stem cell gives rise to all epithelial cells of the gastric gland (Karam 1995). Rats maintain a gastric pH of ≈3.8 (≈3.6 in males and ≈4 in females) which is considerably higher (≈3 times) than the pH value in humans (Iatropoulos 1986), making the rat a more sensitive species, with higher gastric pH, than humans. When a profound general atrophy of the gastric mucosa is produced, the rat responds maximally by hypergastrinemia (Williams, Iatropoulos, and Enzmann 2008). Although gastrin has general trophic effects on the oxyntic cell population of the gastric mucosa (86% in rats and 81% in humans), the main trophic target is the ECL cell population.
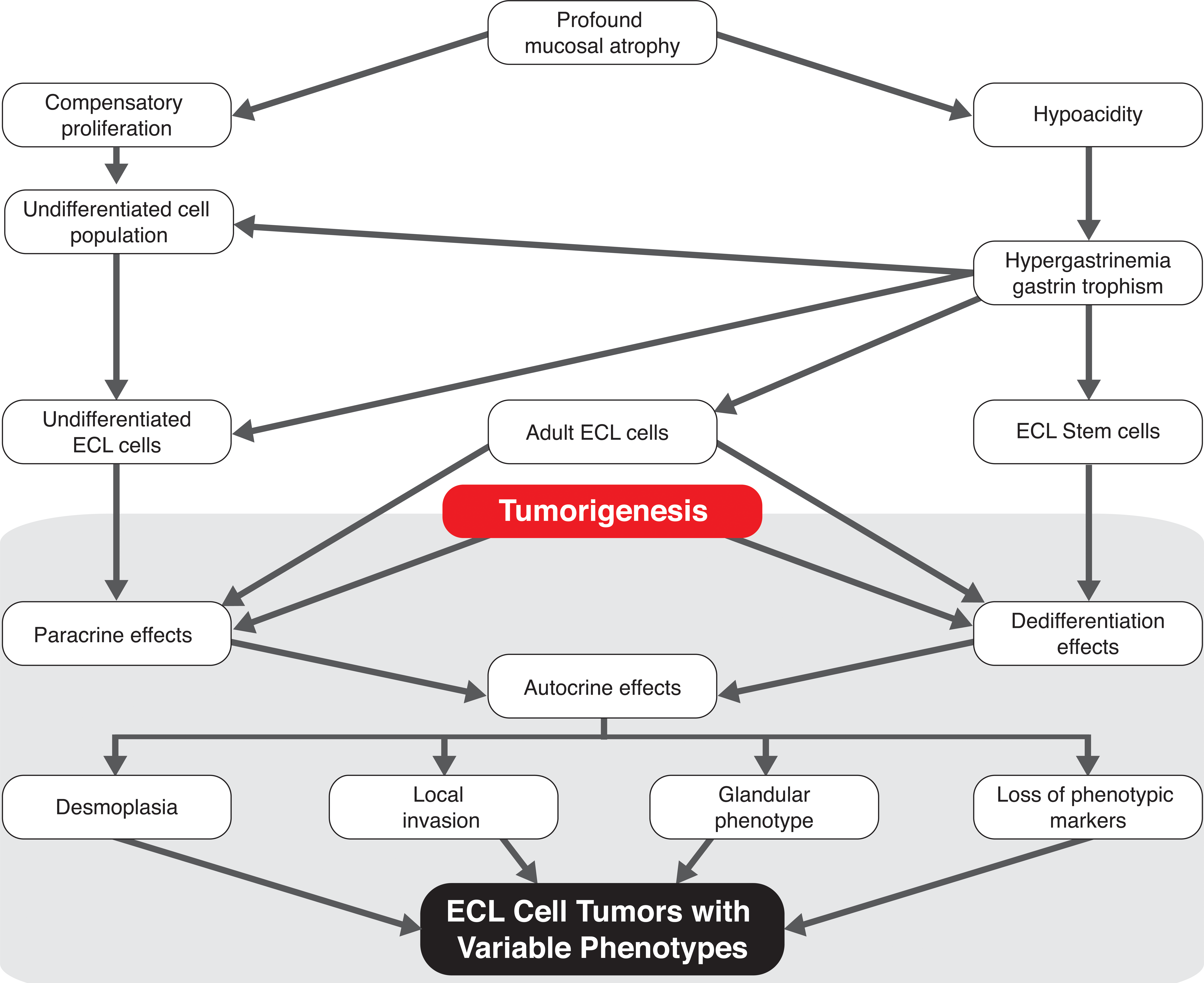
The tumors may result from gastrin trophism that exerts its action on the multipotent stem cells to produce cells that differentiate as ECL cells, it may act on the differentiating ECL cell population or it may act directly on mature ECL cells stimulating them to proliferate as they have the capacity to do (Figure 12). It is logical that a portion of the proliferating cells will be in some stage of differentiation to ECL cells. It may also be that this committed but not fully differentiated population of ECL cells is the primary target of gastrin stimulation which has been previously suggested (Wroblewski et al. 2002). However, irrespective of which cell type/types (i.e., stem cells, differentiating ECL cells, or mature ECL cells) is the target of gastrin stimulation that results in the tumors, it is clear that these gastric tumors originate in the ECL cell population with varying phenotypes having resulted from a desmoplastic response, or by dedifferentiation, both of which have substantial literature documentation as features of ECL cell tumors. Therefore, it is believed that the gastric tumors that develop as a result of long-term treatment with alachlor and butachlor arise from ECL cells by a nongenotoxic mechanism (Hard 2003), involving key events listed in Table 10 and more thoroughly depicted in Figure 12.
Spontaneous ECL Cell Gastric Tumors Observed in Cotton Rats; Correlation with Chloroacetanilide-induced ECL Cell Tumors
The occurrence of spontaneous ECL cell tumors is well known in cotton rats, as a result of sustained hypergastrinemia resulting from hypoacidity (Martinsen et al. 2003). In cotton rats, cancerous ECL cell growth begins as hyperplasia which progresses to dysplasia and with sustained hypergastrinemia results in progression of dysplastic cells to ECL cell gastric tumors. Desmoplasia is often a feature of the ECL cell tumors which grow into the musculature but rarely metastasize (Martinsen et al. 2003), a pattern commonly present in the alachlor and butachlor gastric tumors. In addition to phenotypic similarities, the spontaneous tumors of cotton rats primarily develop in females (Martinsen et al. 2003). It is of interest to mention here that in these female cotton rats, when YF476 (a gastrin receptor antagonist) is given, the tumor-bearing rats displayed thickening of the entire oxyntic mucosa (Martinsen et al. 2003). A female preponderance of gastric ECL cell tumors has also been recorded for ECL cell tumors in humans (Bordi et al. 1986; Naugler and Hunt 2006). This gender difference reflects physiologic differences and subsequent feedback response to gastric pH (Chen et al. 1994; Greaves 2007; Hakanson, Chen, and Sundler 1994; Hakanson et al. 1986; Havu 1986; Williams, Iatropoulos, and Enzmann 2008), as well as a higher population density of ECL cells in females than in males (Havu 1986). Rats in general have a higher density of ECL cells, achieve high levels of gastrin (>100 pg/ml), and are very responsive to elevations of gastrin (Tuch et al. 1992) when compared to humans (Modlin and Sachs 2004; Thake, Hotz, et al. 1995; Thake, Iatropoulos, et al. 1995; Williams, Iatropoulos, and Enzmann 2008), providing further support that this MOA is not relevant for humans. ECL cell tumors apparently dedifferentiate as they grow (Fossmark et al. 2005). This is particularly relevant since in cotton rats the ECL cell tumors can have an adenocarcinoma phenotype with neuroendocrine differentiation (Fossmark et al. 2004; Fossmark, Qvigstad, and Waldum 2008) and ECL cell tumors with similar characteristics can also be induced in cotton rats with the H2 receptor blocker, loxtidine (Fossmark et al. 2004; Modlin and Kidd 2003). A glandular stomach tumor phenotype was also described in CD rats after long-term administration of loxtidine (Poynter et al. 1985).
ECL Cell Gastric Tumors Induced by H2 Receptor Antagonists Blockers (H2RAs) and Proton Pump Inhibitors (PPIs); Correlation with Chloroacetanilide-induced ECL Cell Tumors
It would be expected that gastrin would act on both mature and immature ECL cell populations (Waldum, Brenna, and Sandvik 1998), which would result in an enhanced potential to produce tumors of ECL cell origin. This may explain the differences in phenotypic expression of ECL cell tumors that are associated with alachlor and butachlor as compared to those induced by gastric acid inhibitors, that is, H2RAs and PPIs, since the gastric acid inhibitors do not induce the profound mucosal atrophy, loss of parietal cells, or sustained cell proliferation in the gastric mucosa. Since there is profound mucosal atrophy that precedes tumor formation in alachlor- and butachlor-induced gastric tumors the gastrin acts on a mucosa that is rapidly proliferating in compensation for the loss of glandular (but not surface) epithelium. Therefore, it may have trophic action on multipotential stem cells, on committed but not fully differentiated ECL cells, or on mature ECL cells (Figure 12). The action of gastrin would likely extend to different populations of ECL cells in the chloroacetanilide tumors as compared to the ECL cell tumors that develop as a result of acid blocking substances alone for which there is no glandular atrophic component and therefore no accelerated glandular mucosal cell proliferation. This may explain why dedifferentiation, pleomorphism, anaplasia, and desmoplasia are features of chloroacetanilide tumors, but are much less common in those tumors that develop in rats exposed to H2 receptor inhibitors and PPIs.
Conclusion
Therefore, the Panel concluded that all gastric tumors induced in long-term cancer bioassays and MOA studies performed with alachlor and butachlor were derived from ECL cells at various stages of differentiation, and the process begins with mucosal atrophy (including loss of parietal cells) leading to hypoacidity, hypergastrinemia, sustained ECL cell proliferation, and ECL cell neoplasia. Their end-stage morphology is consistent with known ECL cell tumor morphology and results from dedifferentiation, desmoplasia, and features that are consistent with ECL cell paracrine effects (Figure 12). Based on all available evidence, and consistent with the human relevance framework (Meek et al. 2003), the Panel concluded that chloroacetanilide-induced gastric neoplasia develops via a nongenotoxic MOA, which is subject to a threshold for ECL cell carcinogenicity, and under conditions that are not relevant to humans at known or anticipated levels of exposure.
Footnotes
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Drs. Furukawa and Sherman work for companies that manufacture and/or distribute alachlor and butachlor, and Drs. Harada, Thake, and Iatropoulos were paid consultants.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Acknowledgments
We wish to thank Hiroo Wakimori and Hajime Igarashi for their organizational support and hosting the panel review, Toshiaki Kitazawa, Seigo Hayashi, and Masayoshi Abe for their histopathology support, and Elizabeth G. Webb for her help in preparing the manuscript.