
Abstract
Biotherapeutics are expanding the arsenal of therapeutics available for treating and preventing disease. Although initially thought to have limited side effects due to the specificity of their binding, these drugs have now been shown to have potential for adverse drug reactions including effects on peripheral blood cell counts or function. Hematotoxicity caused by a biotherapeutic can be directly related to the activity of the biotherapeutic or can be indirect and due to autoimmunity, biological cascades, antidrug antibodies, or other immune system responses. Biotherapeutics can cause hematotoxicity primarily as a result of cellular activation, cytotoxicity, drug-dependent and independent immune responses, and sequelae from initiating cytokine and complement cascades. The underlying pathogenesis of biotherapeutic-induced hematotoxicity often is poorly understood. Nonclinical studies have generally predicted clinical hematotoxicity for recombinant cytokines and growth factors. However, most hematologic liabilities of biotherapeutics are not based on drug class but are species specific, immune-mediated, and of low incidence. Despite the potential for unexpected hematologic toxicity, the risk–benefit profile of most biotherapeutics is favorable; hematologic effects are readily monitorable and managed by dose modification, drug withdrawal, and/or therapeutic intervention. This article reviews examples of biotherapeutics that have unexpected hematotoxicity in nonclinical or clinical studies.
Keywords
1. Introduction
Biotherapeutics are expanding the arsenal of therapeutics available for treating and preventing disease. Biotherapeutics are products derived from natural or genetically engineered systems and include monoclonal antibodies (mAbs), recombinant proteins, cytokines, hormones, and nucleic acids. In contrast to small molecules, biotherapeutics (particularly mAbs) are considered to be exquisitely selective for the intended target. Although initially thought to have limited potential for side effects due to the specificity of their binding, a few biotherapeutics have been shown to cause unexpected adverse drug reactions (ADRs) including effects on peripheral blood cell counts or function (Hansel et al. 2010; Pichler 2006). The classes of biotherapeutics associated with unexpected hematologic liabilities include several modalities, including mAbs, cytokines, and hematopoietic growth factors. A meta-analysis of safety reporting databases from 1995 to 2008 estimated that biotherapeutic-associated ADRs of the “blood and lymphatic system” was 1.4-fold that of small molecule pharmaceuticals. However, this estimate included on-target effects, and several biotherapeutics (infliximab, etanercept, adalimumab, and interferon-β1a) were overrepresented in the database (Giezen et al. 2010). In a survey of oncologic biotherapeutics, hematologic toxicity is second only to infusion reactions in terms of the most frequent side effects (Klastersky 2006). To the authors’ knowledge, unexpected nonclinical and clinical hematologic effects of biotherapeutics have not been comprehensively reviewed.
Drug-related hematotoxicity occurs with both small molecule and biotherapeutic pharmaceuticals and generally manifests as decreased circulating blood cell counts. The causes of hematotoxicity include effects on hematopoiesis and blood cell survival, function, and trafficking. Clinically, decreases in cell counts below the lower limit of the reference interval (cytopenia) are graded according to Common Terminology Criteria for Adverse Events (CTCAE) published by the National Cancer Institute of the United States (Table 1). In nonclinical studies, no consensus grades for hematotoxicity exist. Instead, blood cell decreases are attributed to the drug based on evaluation of differences from baseline values and/or a contemporaneous vehicle control group. For the purposes of this review, effects observed in nonclinical species are considered in sections relating to the type of cytopenia (i.e., thrombocytopenia, anemia, neutropenia, etc.), although in some nonclinical examples blood cell counts were not decreased below the historical reference intervals.
CTCAEv4.0 Severity grades for cytopenia (common terminology criteria for adverse events [CTCAEs]; Cancer Therapy Evaluation Program 2012).
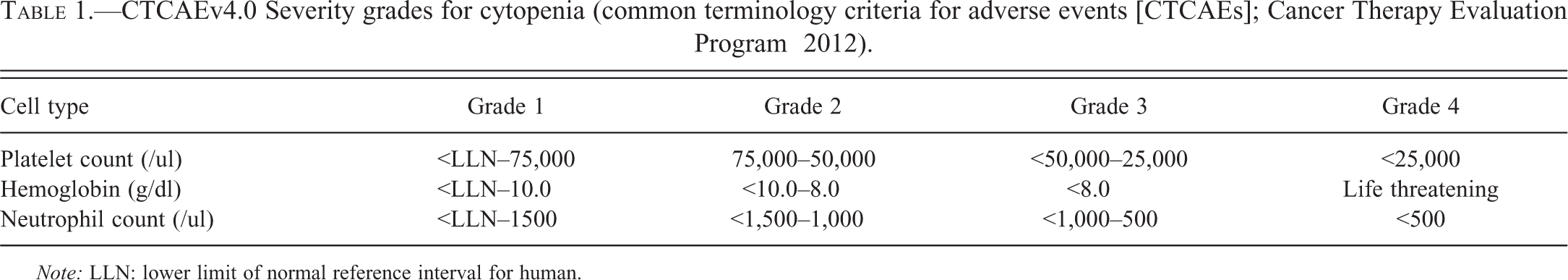
Note: LLN: lower limit of normal reference interval for human.
Hematotoxicity of small molecule pharmaceuticals is extensively documented and may be predictable and related to dose, or idiosyncratic with no dose–response relationship. Small molecule-induced hematotoxicity that is dose related is often translatable from nonclinical research, with high concordance of in vitro and in vivo animal and human studies (Olson et al. 2000; Pessina et al. 2003, 2002). Idiosyncratic hematotoxicity of small molecules (e.g., antipsychotic drugs associated with neutropenia) is less common than predictable toxicity. The underlying cause of idiosyncratic hematotoxicity is unknown, although immune mechanisms are thought to be involved (Aster 2010).
Nonclinical toxicity studies are designed to identify target organ toxicities and predict safety of new drug candidates for humans. The detection of hematological effects in nonclinical species is influenced by intrinsic species characteristics including blood cell life span, metabolic pathways, receptor expression, and the variability of hematologic parameters between individuals (i.e., interindividual variability is lower in rats compared to nonhuman primates; Hall and Everds 2007). Nonclinical testing often predicts high-incidence hematologic effects on conserved processes (e.g., chemotherapeutics affecting rapidly dividing cells in bone marrow) for both small molecules and biotherapeutics. However, nonclinical safety studies are not predictive across species for low-incidence or idiosyncratic hematotoxicities and high-incidence, species-specific effects. Part of the lack of predictivity is due to the small number of animals in nonclinical studies, which limit the ability to detect rare or idiosyncratic effects, compared to the large number of patients dosed in clinical trials. Additionally, the underlying mechanisms for idiosyncratic drug-related hematotoxicity are poorly understood, which impedes the development of relevant animal models (e.g., Uetrecht 1992).
Hematotoxicity can be directly related to the activity of the biotherapeutic or can be indirect and due to autoimmunity, biological cascades, antidrug antibodies (ADA), or other immune system responses (Table 2). In addition, patient factors (including preexisting disease, concurrent medications, and genetics) can influence the occurrence of hematotoxicity. The pathogenesis of hematologic effects of biotherapeutics described in this review is on-target but unexpected, off-target due to interactions with molecules unrelated to the intended pharmacologic target, or poorly understood. Effects of biotherapeutics on erythrocytes, neutrophils, monocyte–macrophages, and platelets, including nonclinical and clinical data are the focus of this review (Table 3). Selected biotherapeutics are discussed that illustrate important pathophysiologic mechanisms and/or have significant nonclinical or clinical hematotoxicity. In addition, biotherapeutic-induced pathophysiologic processes resulting in secondary cytopenias are discussed.
Mechanisms of biotherapeutic-induced hematotoxicity.

Selected biotherapeutics associated with unexpected hematologic effects.
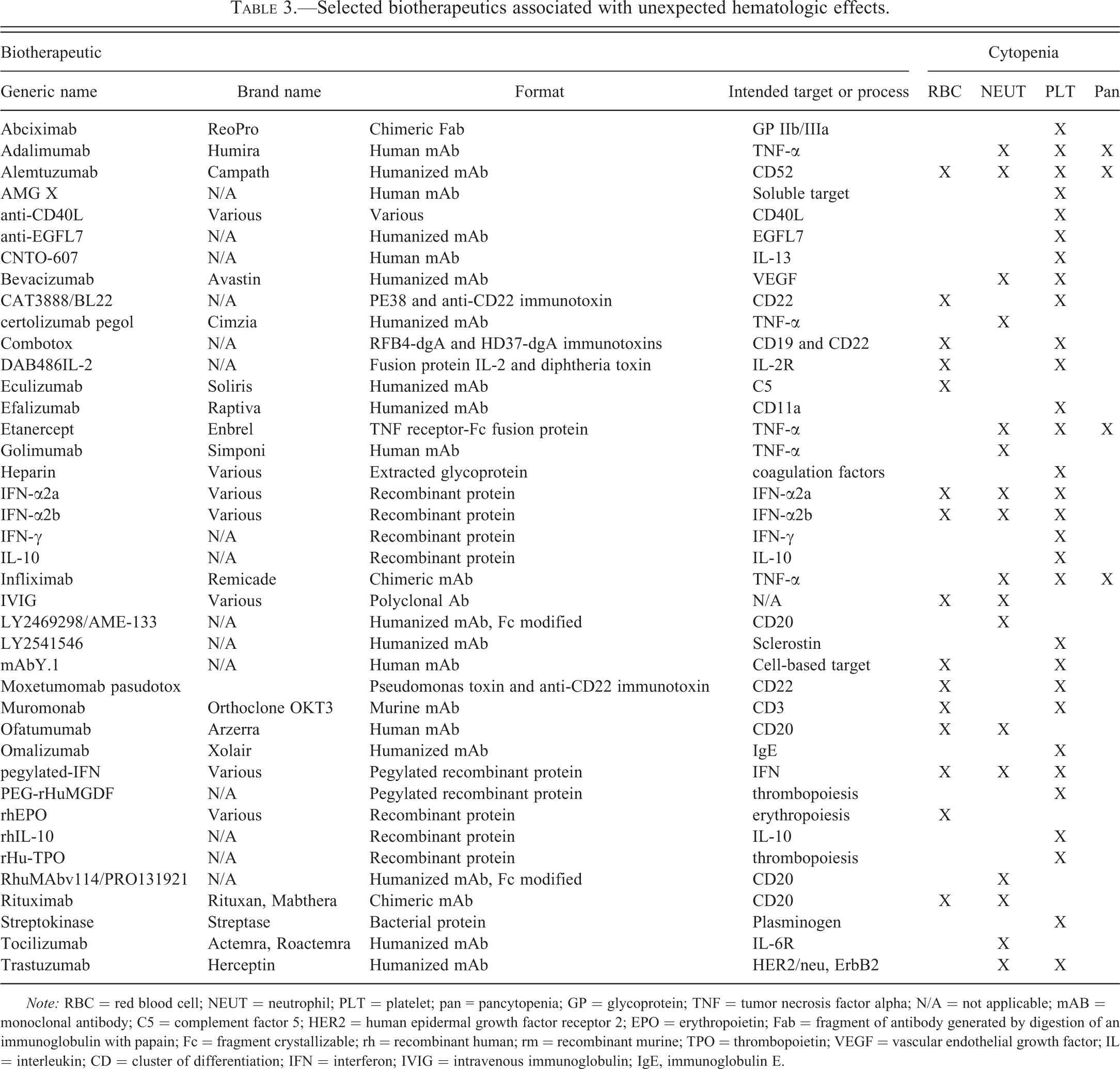
Note: RBC = red blood cell; NEUT = neutrophil; PLT = platelet; pan = pancytopenia; GP = glycoprotein; TNF = tumor necrosis factor alpha; N/A = not applicable; mAB = monoclonal antibody; C5 = complement factor 5; HER2 = human epidermal growth factor receptor 2; EPO = erythropoietin; Fab = fragment of antibody generated by digestion of an immunoglobulin with papain; Fc = fragment crystallizable; rh = recombinant human; rm = recombinant murine; TPO = thrombopoietin; VEGF = vascular endothelial growth factor; IL = interleukin; CD = cluster of differentiation; IFN = interferon; IVIG = intravenous immunoglobulin; IgE, immunoglobulin E.
2. Biotherapeutic-induced Thrombocytopenia
Thrombocytopenia is the most commonly observed unexpected hematologic effect among biotherapeutics based on this review of current nonclinical and clinical literature (Table 4). One potential reason for the overrepresentation of thrombocytopenia in hematotoxicity caused by biotherapeutics is that the platelet surface contains the largest circulating pool of the activating immunoglobulin G (IgG) receptor FcγRIIa (∼5,000 copies/platelet and 200,000–600,000 platelets/µl blood; Worth et al. 2006). This receptor has a low affinity for monomeric IgG but high affinity for complexed Fcγ domains. Cross-linking of FcγRIIa by a biotherapeutic or by ADAs produced as a result of a biotherapeutic can result in platelet activation (Anderson and Anderson 1990; Clemetson and Clemetson 2007).
Selected biotherapeutics associated with thrombocytopenia.a

a Decreased platelet count in nonclinical species may still be within reference interval.
Biotherapeutics can cause thrombocytopenia by decreased production of platelets, shortened platelet life span, or sequestration in vascular beds (Visentin and Liu 2007). Platelet production can be decreased by reduced numbers or altered maturation of megakaryocytes or ineffective production of platelets by mature megakaryocytes. Shortened platelet life span can be due to platelet destruction, or activation, aggregation, and consumption (Aster and Bougie 2007), and is the most common biotherapeutic-related mechanism of thrombocytopenia. Underlying causes include binding of the biotherapeutic to the platelet surface or induction of antiplatelet antibodies that lead to platelet destruction. Additionally, shortened platelet life span can occur through interactions with ADAs that cross-link surface proteins causing platelet activation. Biotherapeutic-induced hemophagocytosis, complement activation, cytokine release, hypersensitivity reactions, and thrombosis can also shorten platelet life span and lead to decreased platelet counts.
The underlying cause of thrombocytopenia determines the rate of recovery; effects on earlier processes (e.g., megakaryocyte and platelet production) tend to recover slower than effects on platelet life span or sequestration. In humans and laboratory animals, platelet counts greater than 50,000/µl are considered sufficient for hemostasis, while counts <10,000/µl are associated with an increased risk of hemorrhage (Russell 2010). Clinical grading categories for thrombocytopenia are listed in Table 1.
2.1. Multimeric Complexes Localized to Platelets
The localization of multimeric complexes of antibodies (mAbs or endogenous antibodies) with multivalent endogenous antigens on the surface of platelets predisposes subjects to the development of thrombocytopenia, sometimes with thrombosis. This mechanism is responsible for thrombocytopenia and thrombosis due to heparin and streptokinase and postulated for anti-CD40L molecules and bevacizumab.
2.1.1. Heparin
Heparin is an antithrombotic biotherapeutic prepared from the mucosa of domestic animals (porcine intestine or bovine lung). Heparin administration to humans is associated with two types of thrombocytopenia: nonimmune heparine–associated thrombocytopenia and heparin-induced thrombocytopenia (HIT; Shantsila et al. 2009).
Nonimmune heparin–associated thrombocytopenia (formerly called type I HIT) occurs in approximately 25% of patients within a few days of dosing with heparin and is characterized by a mild decrease in platelets that resolves despite continued treatment. It is thought to be caused by inhibition of adenylate cyclase resulting in in vivo platelet aggregates and is not associated with a prothrombotic state (Chong 1995 reviewed in Chong 2007).
HIT (formerly called type II HIT; reviewed in Shantsila et al. 2009) usually occurs within 1 to 2 weeks after first exposure or within hours of a repeat exposure in 1 to 3% of patients receiving unfractionated heparin and in 0 to 0.8% of patients receiving low-molecular-weight heparin. The condition is characterized by moderate to profound thrombocytopenia and a prothrombotic state. About 25% of patients who develop thrombocytopenia will also develop thrombosis primarily in large arteries and veins (Walenga, Jeske, and Messmore 2000). HIT is caused by antibodies that bind to heparin complexed with platelet factor 4 (PF4) on the platelet surface, but not all patients that develop antibodies to heparin proceed to develop HIT.
Under normal conditions, PF4 stored in the alpha granules of platelets is released upon platelet activation. Administration of heparin also causes increased plasma PF4 by displacing endothelial-associated PF4 (Visentin et al. 1994). PF4 from activated platelets or endothelium can bind to platelet-surface molecules such as glycosaminoglycans and heparin (Greinacher 2009; Greinacher et al. 1994; Newman and Chong 2000). Heparin administered as a therapeutic can also bind to circulating PF4 or PF4 localized on the surface of platelets. Heterogeneous antibodies (HIT antibodies) develop in response to newly exposed cryptic epitopes or neoantigens formed when heparin binds to PF4. These HIT antibodies and heparin/PF4 complexes form multimeric structures that activate platelets via FcγRIIa receptors. This results in degranulation and release of procoagulant substances, further activation of platelets, and release of more PF4. Activated platelets are removed from circulation by macrophages or are consumed in the formation of thrombi. HIT antibodies also activate monocytes and endothelial cells by a similar mechanism. Together, these processes contribute to a procoagulant state that can result in arterial and venous thromboses. In patients with HIT antibodies without thrombocytopenia or thrombosis, the monocyte–macrophage system is presumed to clear HIT antibody-coated platelets without platelet activation.
HIT has been reported in nonclinical studies in rats, pigs, and rabbits.
2.1.2. Streptokinase
Streptokinase (Streptase) is a bacteria-derived thrombolytic agent. In rare instances, administration of streptokinase is associated with clot propagation (Fitzgerald et al. 1988; Goldsmith, Lollar, and Hoak 1981). The cause of paradoxical clot propagation in response to streptokinase therapy is thought to be due to platelet activation caused by anti-streptokinase antibodies in a subset of patients with these antibodies. ADAs bind to the streptokinase–plasminogen complex on the surface of platelets, releasing procoagulant substances (Vaughan et al. 1991). Platelet activation by streptokinase requires cleavage of protease-activated receptor-1 (PAR-1) on the surface of platelets as well as interaction of endogenous anti-streptokinase antibodies with the FcγRIIa on the surface of platelets (McRedmond et al. 2000). Thus, streptokinase-induced platelet activation shares some of the characteristics of HIT, as ADAs form higher-order complexes with the drug on the surface of platelets, resulting in platelet activation. To the authors’ knowledge, platelet activation due to anti-streptokinase antibodies has not been reported in nonclinical species.
2.1.3. Anti-CD40L mAbs
Several mAb biotherapeutics have been developed against CD40 ligand (CD40L). Both CD40 and CD40L are expressed on activated T-cells and activated platelets (Danese and Fiocchi 2005). The development of anti-CD40L therapeutics was discontinued due to a high incidence of thrombocytopenia and thrombosis in nonhuman primates and humans (Boumpas et al. 2003; Kawai et al. 2000; Schuler et al. 2004). Two mechanisms are thought to contribute to thrombosis by anti-CD40L mAbs. Anti-CD40L mAbs can inhibit disaggregation of ADP-aggregated platelets in vitro, which may explain the mechanism of thromboemboli associated with anti-CD40L mAbs (Mirabet et al. 2008). Additionally, because CD40L is a trimeric structure, multiple mAbs targeting CD40L can bind and form higher-order immune complexes on the platelet surface, in a process similar to that of HIT antibodies (Hatfield et al. 2011; Robles-Carrillo et al. 2010). Similar findings of thrombosis were not observed in nonclinical rodent models, presumably because they lack FcγRIIa receptors (Robles-Carrillo et al. 2010).
2.1.4. Bevacizumab
Bevacizumab (Avastin) is a humanized mAb against vascular endothelial growth factor (VEGF), a protein that stimulates angiogenesis. Bevacizumab administration has been associated with thrombocytopenia and thrombosis in human trials but not nonclinical safety studies (Leal and Robins 2010; Scappaticci et al. 2007). Postulated mechanisms include platelet consumption due to endothelial dysfunction from lack of VEGF (Kilickap, Abali, and Celik 2003), a HIT-like mechanism, and direct effects on platelet life span. VEGF is highly enriched in platelets and, upon release, can bind to heparin on the platelet surface (Kut, Mac Gabhann, and Popel 2007; Peterson et al. 2010; Verheul et al. 1997). Binding of multiple bevacizumab molecules to the VEGF/heparin complex may result in higher-order immune complexes on the surface of platelets, which may activate platelets causing thrombocytopenia and thrombosis. In support of this hypothesis, genetically engineered mice that express human FcγRIIa on platelets developed thrombocytopenia and thrombosis when hypercoagulable conditions were simulated and subtherapeutic concentrations of heparin were coadministered with bevacizumab (Meyer et al. 2009). In addition, shortened platelet life span has been reported in platelets that internalize bevacizumab (Verheul et al. 2007).
2.2. Induction of Antiplatelet Antibodies
2.2.1. Abciximab
Abciximab (ReoPro) is a Fab fragment of a chimeric mAb against the platelet fibrinogen receptor GPIIb/IIIa. Administration of abciximab causes mild to severe thrombocytopenia in approximately 0.5 to 5% of human patients after the first dose (Aster 2007; Curtis et al. 2002; Said et al. 2007). The overall incidence and severity of thrombocytopenia increase with reexposure to abciximab, likely due to higher ADA titers (Berkowitz et al. 1997; Dery et al. 2004). Although three marketed anti-GPIIb/IIIa drugs (abciximab, and the small molecule drugs tirofiban and eptifibatide) are associated with thrombocytopenia in humans, patients that have experienced thrombocytopenia with one GPIIb/IIIa inhibitor may not be at increased risk for thrombocytopenia with other GPIIb/IIIa inhibitors (Patel et al. 2005; Rao and Mascarenhas 2001; Tcheng 2000).
Onset of thrombocytopenia usually occurs rapidly (within hours) after the first dose but can be delayed up to 5 to 10 days (Aster 2007; Reddy, Carmody, and Kereiakes 2001). Thrombocytopenia can be severe (platelet counts <50,000/µl) to profound (platelet counts <20,000/µl), but the risk of bleeding is higher than the platelet counts would suggest because of the inhibitory effect of the drug on platelet function. Abciximab-associated thrombocytopenia has not been reported in standard nonclinical animal species including macaques, although thrombocytopenia associated with other anti-GPIIb/IIIa molecules has been reported in baboons (Tanaka et al. 2005).
The underlying cause of abciximab-related thrombocytopenia is thought to be preexisting or newly formed antibodies, generally directed against the murine sequences of chimeric Fab molecule (Curtis et al. 2002; Said et al. 2007). The interaction of abciximab with GPIIb/IIIa may also expose neoantigens. Abciximab may induce the formation of ADAs that activate platelets directly (Lajus et al. 2010). The localization of the drug to GPIIb/IIIa on platelets and the binding of ADAs to the drug results in immune-mediated removal of platelets.
2.2.2. Tumor Necrosis Factor Alpha (TNF-α) Inhibitors
TNF-α inhibitors block the activity of the cytokine TNF-α and include adalimumab (Humira), a fully human mAb to TNF-α, infliximab (Remicade), a humanized mouse mAb to TNF-α, and etanercept (Enbrel), a TNF-α receptor2-Fc fusion protein. TNF-α inhibitors, either alone or in combination with IFNγ, have been linked to mild to severe (approximately CTCAE grades 3–4) thrombocytopenia in up to 6% of patients (Brunasso and Massone 2009; Chen et al. 2011; Gerloni et al. 2008; Michelmann et al. 1997; Nurnberger et al. 1994; Pathare, Heycock, and Hamilton 2006; Salar et al. 2007). Similar findings were not reported in nonclinical studies.
Thrombocytopenia associated with TNF-α inhibitors can occur at any time during treatment, from less than 1 day to 3 years (Brunasso and Massone 2009). Cessation of dosing is often associated with clinical improvement, while reexposure to the drug results in relapse. Exposure to a second TNF-α inhibitor is inconsistently associated with recurrent thrombocytopenia (Brunasso and Massone 2009; Gerloni et al. 2008; Pathare, Heycock, and Hamilton 2006; Salar et al. 2007). The relative risk for thrombocytopenia among the three commonly used TNF-α inhibitors is uncertain, although infliximab may be overrepresented (Brunasso and Massone 2009). The increased propensity for immunogenicity of infliximab may contribute to the higher relative incidence of thrombocytopenia (5.2. Infusion Reactions; Brunasso and Massone 2009; Chen et al. 2011).
Two primary mechanisms have been proposed for thrombocytopenia associated with TNF-α inhibitors. TNF-α inhibitors commonly induce autoantibodies including antiplatelet antibodies (Brunasso and Massone 2009; Caramaschi et al. 2009; Pathare, Heycock, and Hamilton 2006; Salar et al. 2007). Autoantibody generation is possibly related to TNF pharmacology as TNF normally inhibits T-cell responses that suppress autoreactive B cells (Via et al. 2001). However, the incidence of autoimmune cytopenia (thrombocytopenia and/or neutropenia) is low compared to the incidence of autoantibody production. Antiplatelet antibodies induced by TNF-α inhibitors can generally bind to platelets in the absence of drug (drug-independent antibodies). Additionally, because TNF-α plays a complex role in hematopoiesis, suppression of hematopoiesis by TNF-α inhibitor therapy may contribute to thrombocytopenia (Keystone 2001; Pathare, Heycock, and Hamilton 2006).
The risk of thrombosis due to TNF-α inhibitor therapy and its relationship to thrombocytopenia is uncertain. A review of records from a 7-year period of 85 patients on TNF-α inhibitor therapy that developed arterial and/or venous thrombosis led these authors to suggest that the therapy may be responsible for increased thrombosis (Petitpain et al. 2009). In contrast, patients with rheumatoid arthritis treated with TNF-α inhibitor therapies had no increase in venous thrombotic events in a recent large study of over 15,000 patients (Davies et al. 2011).
2.2.3. Interferon Alpha
Interferon alpha (IFN-α) is a recombinant cytokine that is marketed as several different forms including IFN-α2a or -2b and pegylated (peg)-IFN-α2a or -2b. Treatment of patients with all forms of IFN-α, with or without cotreatment with the antiviral ribavirin, is associated with mild (grades 2–3) or severe (grade 4) thrombocytopenia (Elefsiniotis et al. 2006; Kim et al. 2010; Li, Han, and Lu 2010; Sagir et al. 2002; Sevastianos et al. 2003; Yang et al. 2010). Thrombocytopenia associated with IFN-α therapy occurs most frequently in hepatitis C patients (Dourakis, Deutsch, and Hadziyannis 1996; Fujii et al. 2003; Kim et al. 2010; Li, Han, and Lu 2010; Medeiros et al. 2004; Sevastianos et al. 2003) and is also reported in oncology patients (Abdi, Brien, and Venner 1986; Abdi and Venner 1986; Akamatsu et al. 2006; Demirturk et al. 2006; McLaughlin et al. 1985; Zuffa et al. 1996). Because diseases treated with IFN-α are also those that predispose patients to thrombocytopenia, the direct relationship of thrombocytopenia to IFN-α therapy can be difficult to elucidate (Ramos-Casals et al. 2003). Thrombocytopenia was also observed in cynomolgus and rhesus monkeys in nonclinical studies with IFN-α therapy (PEG-INTRON [Peg-IFN-α2b] Toxicologists Review 2004; Pegasys [Peg-IFN-α2a] Preclinical Review 2009; and Peginterferon alpha 2a, Scientific Discussion 2005).
The mild form of IFN-α-related thrombocytopenia is most common and occurs in approximately 23% of patients (Alvarez et al. 2011; Ernstoff and Kirkwood 1984). Mild thrombocytopenia associated with IFN-α is caused by decreased platelet production, based on the finding of normal or slightly decreased bone marrow megakaryocyte cellularity (Ernstoff and Kirkwood 1984; Sata et al. 1997; Wadenvik et al. 1991). IFN-α may inhibit platelet production by suppressing the maturation of megakaryocytic demarcation membranes required for platelet release (Yamane et al. 2008). Slightly decreased platelet survival or redistribution may also contribute to thrombocytopenia (Sata et al. 1997; Wadenvik et al. 1991).
The clinical course of severe thrombocytopenia with IFN-α, occurring in less than 1% of patients, is generally acute, with patients presenting with epistaxis, ecchymoses, petechiae, or other signs of altered hemostasis. The time of onset is variable, occurring between 1 and 36 months after the start of IFN-α therapy, and usually resolves with cessation of dosing (Li, Han, and Lu 2010; McLaughlin et al. 1985). The mechanism for severe thrombocytopenia associated with IFN-α is immune-mediated, with clinicopathologic findings of platelet-associated antibodies, megakaryocytic hyperplasia, and clinical response to treatment withdrawal and administration of corticosteroids (Enomoto et al. 2008; Fujii et al. 2003; Lambotte et al. 2005; Zuffa et al. 1996). Patients have been successfully dosed with a different IFN-α biotherapeutic, with no recurrence of thrombocytopenia (Akamatsu et al. 2006; Arimura et al. 2004). Specific patterns of single-nucleotide polymorphisms (SNPs) involving the DDRGK1 and ITPA (inosine triphosphate pyrophosphatase) genes show an association with immune-mediated thrombocytopenia in hepatitis C patients treated with peg-IFN-α2a (Tanaka et al. 2011; Thompson et al. 2012).
IFN-α therapy is also associated with thrombocytopenia secondary to thrombotic thrombocytopenic purpura (TTP; Pisoni, Ruggenenti, and Remuzzi 2001); this condition is discussed with thrombotic microangiopathies (TMAs) in Section 5.3.
2.2.4. Efalizumab
Efalizumab (Raptiva) is a humanized mAb against CD11a. Thrombocytopenia (platelet counts ≤52,000/µl) was reported in 6 (0.22%) of 2,762 human patients treated with efalizumab (Raptiva prescribing information 2005; Warkentin and Kwon 2005). The onset of clinical signs of altered hemostasis is variable (from 1 day to 3 years, but usually between 2 and 12 weeks after therapy; Hostetler, Zirwas, and Bechtel 2007; Tom et al. 2006; Warkentin and Kwon 2005). Discontinuation of efalizumab and treatment with corticosteroids resolves most cases of thrombocytopenia. Based on the generally rapid response to corticosteroid therapy, efalizumab-related thrombocytopenia is considered to be immune mediated (Warkentin and Kwon 2005). To the authors’ knowledge, thrombocytopenia has not been reported with efalizumab in nonclinical studies.
2.2.5. Alemtuzumab
Alemtuzumab (Campath) is an anti-CD52 human mAb that targets mature circulating lymphocytes. In clinical trials, alemtuzumab caused immune thrombocytopenia in 2.8% of patients with relapsing-remitting multiple sclerosis (Cuker 2011; Fogarty 2011). Unlike most drug-induced thrombocytopenia that occurs within days of exposure to a drug, thrombocytopenia with alemtuzumab occurred at a median of 10.5 months after the last exposure to the drug. Most of these patients recovered rapidly from thrombocytopenia with conventional therapy (glucocorticoids, IVIG, platelet transfusion), unlike adult patients with idiopathic immune thrombocytopenia that generally develop chronic disease. Researchers hypothesized that transient thrombocytopenia may develop during the reconstitution of peripheral blood cells after therapy. Expression of self-reactive lymphocytes producing antiplatelet antibodies may be transient due to deletion of these self-reactive clones during reestablishment of normal lymphoid populations. To the authors’ knowledge, thrombocytopenia has not been reported with alemtuzumab in nonclinical studies.
2.2.6. Trastuzumab
Trastuzumab (Herceptin) is a humanized mAb that targets the human epidermal growth factor receptor 2 (HER2). In rare cases, administration of trastuzumab has been associated with onset of severe (grade 4) thrombocytopenic events within days to weeks of the first dose (Jara Sanchez et al. 2009). In at least three cases, thrombocytopenia resulted in discontinuation of the drug (Cathomas and von Moos 2009; Jara Sanchez et al. 2009; Parikh, Neave, and Palmieri 2008).
The underlying cause of trastuzumab-associated thrombocytopenia has not been determined, but clinical responses to standard therapy for immune mediated thrombocytopenia (intravenous immunoglobulin [IVIG] and glucocorticoids) in most cases have led the investigators to suggest drug-induced immune-mediated thrombocytopenia (Cathomas and von Moos 2009; Drudi et al. 2010; Jara Sanchez et al. 2009; Mantzourani et al. 2011; Parikh, Neave, and Palmieri 2008).
To the authors’ knowledge, thrombocytopenia has not been reported with trastuzumab in nonclinical studies.
2.3. Direct Binding of Biotherapeutic to Platelets and/or Megakaryocytes
2.3.1. AMG X
AMG X is a human mAb that neutralizes a soluble human protein. AMG X caused marked thrombocytopenia (mean platelet count of 21,000/µl at 15 min after a dose of 300 mg/kg IV), platelet activation, transient loss of consciousness, and reduced mean arterial pressure in cynomolgus monkeys at doses ≥15 mg/kg after the first intravenous administration (Santostefano et al. 2012). Other mAbs against the same pharmacological target failed to induce these in vivo effects.
The underlying mechanism was considered to be direct platelet activation, causing release of serotonin and the subsequent cardiovascular effects. In vitro, AMG X induced activation in platelets from multiple macaque species. The target protein was not expressed on macaque platelets, suggesting that platelet activation occurred through an off-target mechanism. AMG X bound directly to platelets of cynomolgus macaques and caused release of serotonin in vitro. Macaque platelet activation required both recognition/binding of a platelet ligand with the Fab domain, and interaction of platelet FcγRIIa receptors with the Fc domain of AMG X. AMG X did not bind to, activate, or cause serotonin release from human platelets in vitro, suggesting that this off-target effect was macaque-specific.
2.3.2. LY2541546
LY2541546 is a humanized mAb against human sclerostin. Weekly dosing of LY2541546 caused thrombocytopenia (nadir not evaluated) associated with clinical signs of altered hemostasis in rats but not in cynomolgus monkeys in nonclinical toxicity studies (Rudmann et al. 2012). The altered hemostasis was considered secondary to thrombocytopenia.
Thrombocytopenia was considered to be an off-target and rat-specific effect of LY2541546 as sclerostin was not expressed in rat, cynomolgus monkey, or human megakaryocytes or platelets. Immunohistochemistry and immunocytochemistry indicated that LY2541546 bound to rat megakaryocytes and platelets but not to those of humans or cynomolgus monkeys. Binding of LY2541546 to megakaryocytes and platelets was thought to result in shortened platelet half-life and possibly ineffective production. Human data have not been reported for this test article.
2.4. Antidrug Antibodies that Cross-React with Endogenous Thrombopoietin
Earlier-generation thrombopoietic drugs had significant amino acid homology compared to natural thrombopoietin (TPO). A low number of patients treated subcutaneously with the thrombopoietic agents rHu-TPO or PEG-rHuMGDF (megakaryocyte growth and development factor) developed ADAs that cross-reacted with TPO. These ADAs neutralized endogenous TPO as well as the biotherapeutic, resulting in severe (grade 4) thrombocytopenia (Basser et al. 2002; Kuter and Begley 2002; Li et al. 2001). Newer thrombopoietic drugs (e.g., romiplostim and eltrombopag) do not include sequences of naturally occurring TPO and thus have not been associated with ADAs that cross-react with endogenous TPO (Cuker 2010).
2.5. Thrombocytopenia of Undetermined Mechanism
2.5.1. Omalizumab
Omalizumab (Xolair) is a humanized mAb against human IgE. Omalizumab caused dose-related, age-dependent, reversible decreases in platelet counts in adult and juvenile (8- to 10-month-old) monkeys (Review of CMC for Xolair 2003). Clinical trial data in addition to a meta-analysis of clinical data found no risk of thrombocytopenia in patients treated with omalizumab (Corren et al. 2009). Safety pharmacovigilance for omalizumab has noted mild (grade 2 or greater) decreases in peripheral blood platelet counts in a few patients, without evidence of bleeding or changes to hemoglobin (Review of CMC for Xolair 2003; Xolair: European Public Assessment Report 2012). The platelet decreases in patients were transient, contrasting with a prolonged decrease in nonhuman primates. In vitro, omalizumab did not bind to or activate human or nonhuman primate platelets and did not inhibit aggregation by commonly used platelet agonists. An underlying cause of platelet decreases in monkeys and humans was not determined.
2.5.2. Interleukin-10
Recombinant human interleukin-10 (IL-10) is a cytokine that consistently causes mild thrombocytopenia in healthy volunteers and patients, but not in nonclinical studies. Platelet counts decreased to a mean nadir of 164,000/µl (60% of pretest values) between 6 and 8 days after a single dose and recovered within a few days after cessation of dosing (Huhn et al. 1997, 1996; Sosman et al. 2000). Humans administered IL-10 also show decreases in splenic sequestration of platelets, which has been proposed as a compensatory reaction to decreased peripheral platelet counts.
The underlying cause of thrombocytopenia caused by IL-10 may be decreased production, based on increased circulating concentration of TPO, decreased numbers of CFU-MK in bone marrow but no effects on platelet survival (Sosman et al. 2000). Decreased platelet production may be related to IL10-mediated regulation of proinflammatory cytokines (i.e., IL-6) involved in megakaryocytopoiesis and thrombopoiesis.
2.5.3. CNTO-607
CNTO-607, a fully human IgG1 mAb targeted to IL-13, caused minimal to moderate decreases (approximately 10–30%) in mean platelet counts in cynomolgus monkeys starting within a day after the first dose (Martin et al. 2008; Personal Communication, P.L. Martin, 2012). Individual animal platelet counts did not decrease below the historical reference interval and were considered nonadverse by the investigators. Platelet counts recovered within 1 to 4 weeks after a single and multiple doses, respectively. The underlying cause of platelet decreases was not determined but may be due to decreased life span of platelets, since megakaryocyte density in the bone marrow was slightly higher in treated animals compared to controls at 4 days after the third dose. Of note, CNTO-607 had undesirable biophysical properties leading to low solubility and drug aggregates (Wu et al. 2010). These characteristics of CNTO-607 could potentially have enhanced Fc-mediated interaction with platelets. Clinical data with CNTO-607 have not been reported.
3. Biotherapeutic-induced Anemia
Decreased red cell mass, sometimes resulting in anemia, is an uncommon adverse drug response to biotherapeutics and can be caused by effects on circulating erythrocytes (hemolysis) or erythropoiesis in the bone marrow (Table 5). Hemolysis can result from generation of drug-induced endogenous antibodies (immune hemolysis); direct binding of the therapeutic antibody to erythrocytes; complement activation; and secondary to platelet activation (discussed in section 5.3. on TMA). Clinical grading categories for anemia are listed in Table 1.
Selected biotherapeutics associated with anemia.a

Note: IFN = interferon; IVIG = intravenous immunoglobulin; rhEPO = recombinant human erythropoietin. aDecreased red cell mass in nonclinical species may be within reference intervals.
Drug-related immune hemolysis is generally due to the de novo development of pathogenic antibodies that bind to red blood cells in the presence or absence of drug (i.e., drug-dependent or independent antierythrocyte antibodies, respectively). Biotherapeutics that induce immune hemolysis generally are associated with drug-independent antibodies. In contrast, drug-dependent antibodies are more common with small molecule drug-induced hemolysis (Garratty and Petz 2007). Binding of the Fab portion of pathogenic antibodies to erythrocytes results in opsonization and extravascular hemolysis through Fc- and/or C3b-mediated phagocytosis by macrophages. Intravascular hemolysis can occur as a result of complement fixation due to pathogenic antibodies or dysregulated complement activation (Garratty and Petz 2007) or via antibody-dependent cytotoxicity (ADCC) mediated by natural killer (NK) cells (Cunningham and Silberstein 2005).
Clinically, immune hemolysis is characterized by decreased red cell mass parameters (hematocrit, hemoglobin, and red cell count) with increased reticulocytes. Additional findings may include decreased serum haptoglobin and increased total and unconjugated bilirubin. The direct antiglobulin test (DAT, a Coombs test) demonstrates the presence of antibody and/or complement associated with the red cell membrane. Indirect antiglobulin tests and hemolytic assays detect pathogenic antibodies in the serum of biotherapeutic-treated subjects by incubation in vitro with erythrocyte suspensions and addition of antitherapeutic or species-specific antiimmunoglobulin with and without complement. Detection of red cell–binding antibody can be further investigated by flow cytometry (to detect immunoglobulin bound to the cell surface), radiolabeling of therapeutic antibody and incubation with erythrocyte suspensions in vitro, and enzyme-linked immunosorbent assay (ELISA) using plate-bound red cell membranes and the biotherapeutic or the serum from biotherpeutic-treated individuals.
Decreased red cell production due to biotherapeutics can occur by interference with erythropoietic cytokines and growth factors, direct cytotoxicity, or drug-related endogenous antibodies that inhibit the development of erythroid cells in bone marrow. If severe enough, these processes can result in pure red cell aplasia (PRCA), a selective absence of erythroid cells in the bone marrow. Diagnosis of an effect on red cell production generally involves enumeration of peripheral blood reticulocytes and, particularly in nonclinical studies, microscopic examination of the bone marrow.
3.1. Induction of Antierythrocyte Antibodies
3.1.1. Alemtuzumab
Alemtuzumab (Campath, MabCampath, Campath 1-H) is a humanized anti-CD52 mAb. Alemtuzumab has been implicated in causing DAT-positive immune hemolysis in humans (Elimelakh et al. 2007), but not in nonclinical studies. In a study of pancreatic transplant recipients given a combination of alemtuzumab, daclizumab, and mycophenolate mofetil, immune hemolysis and/or PRCA occurred in 5.6% (20 of 138) patients (Elimelakh et al. 2007). The anemia had a delayed onset, with a median diagnosis at 9 months (range 2 to 15 months) following cessation of treatment. The alemtuzumab-independent antierythroid antibodies of subjects with more severe anemia fixed complement to a greater degree than those of subjects with milder anemia. The combined immunosuppressive effect of alemtuzumab, daclizumab, and mycophenolate mofetil is suspected to have contributed to immune dysregulation and subsequent immune hemolysis (Elimelakh et al. 2007).
3.1.2. IFN-α
Administration of IFN-α-2a or -2b is associated with rare cases of DAT-positive immune hemolysis in humans, but not in nonclinical studies. IFN-α can cause an exacerbation of autoimmunity due to preexisting anti-red cell antibodies or induction of de novo, drug-independent immune hemolysis (Andriani et al. 1996). In the former, hemolytic anemia occurs after a few days of treatment, while de novo cases of immune hemolysis occur after months of IFN-α treatment (Andriani et al. 1996; Sacchi et al. 1995). Hemolysis is potentially an adverse pharmacologic effect of the biotherapeutic as IFN-α has been shown to suppress T-regulatory lymphocytes and therefore could predispose patients to autoimmune hemolytic anemia (Golding et al. 2010).
3.1.3. Ofatumumab
Ofatumumab (Arzerra) is a human mAb directed against CD20. In nonclinical studies, ofatumumab caused mild to severe, DAT-positive immune hemolysis in cynomolgus macaques (Lemery et al. 2010). Ofatumumab also bound to red cells in a subset of DAT-positive animals. In a clinical trial, anemia occurred in 6.5% of patients (9 of 138), and 2 of these patients were diagnosed with hemolytic anemia (Wierda et al. 2010). The underlying cause of hemolytic anemia was not assessed in these patients, so the relationship of the nonclinical hematotoxicity to the clinical findings is uncertain. Further assessment of the pathogenic antibody in human patients was not reported.
3.2. Direct Binding of Biotherapeutics to Red Blood Cells—Intravenous Immunoglobulin
IVIG is a human polyclonal antibody blood product. Paradoxically, although used as an immunosuppressive treatment for autoimmune disease including autoimmune-mediated hemolytic anemia, IVIG can also induce immune hemolysis in human patients. This is attributed to anti-red cell antibodies contained in the drug product (Baxley and Akhtari 2011). Host factors also contribute to the development of hemolysis, with risk factors including a non-O blood group and preexisting inflammation in the recipient (Padmore 2012). Underlying mechanisms include extravascular hemolysis from opsonized red cells and intravascular hemolysis by complement fixation. In addition to anti-red cell antibodies, aggregated immunoglobulin in IVIG preparations can fix complement, bind to erythrocytes through CR1, and result in hemolysis, particularly in older patients (Kessary-Shoham et al. 1999). There have been no reports of IVIG-induced hemolysis in nonclinical species.
3.3. Pure Red Cell Aplasia from Antidrug Antibodies that Cross-react with Endogenous Erythropoietin
Recombinant human erythropoietin (rhEPO) is a biotherapeutic that stimulates erythropoiesis. In some individuals (humans and other species), ADAs against rhEPO can neutralize both the biotherapeutic and endogenous EPO resulting in PRCA and lead to severe nonregenerative anemia (Cowgill et al. 1998; Eckardt and Casadevall 2003). Human cases have primarily occurred in the setting of treatment for anemia of chronic kidney disease (Eckardt and Casadevall 2003). Unexpectedly increased immunogenicity with older generation rhEPO molecules (predominately Eprex) in non-U.S. markets was associated with human cases of PRCA during a restricted time period (1998–2002) that coincided with a formulation change (Eckardt and Casadevall 2003; McKoy et al. 2008). Potential causes of the increased immunogenicity were formulations that resulted in aggregate formation, leachates from uncoated rubber stoppers acting as adjuvants, poor product handling, and/or subcutaneous administration (McKoy et al. 2008). Changes to manufacturing and product handling have resulted in a much lower incidence of PRCA with currently marketed rhEPO products (McKoy et al. 2008).
3.4. Complement-dependent Extravascular Hemolysis—Eculizumab
Eculizumab (Soliris) is a humanized mAb directed against complement 5 (C5) developed for treatment of paroxysmal nocturnal hemoglobinuria (PNH). PNH is an acquired disease caused by a failure in complement regulation on the red cell surface with subsequent hemolysis and thrombosis. Eculizumab was developed to reduce complement-mediated intravascular hemolysis in PNH patients by neutralization of C5-propagated membrane attack complex on the erythrocyte cell membrane. Although patients with severe PNH benefit from eculizumab therapy, some patients still require transfusions due to eculizumab-related extravascular hemolysis (Risitano et al. 2009). Complement factor 3 (C3), an earlier molecule than C5 in the complement cascade, accumulates on the erythrocyte surface in a low number of patients dosed with eculizumab. The accumulation of C3 on the red cell surface results in opsonization and extravascular hemolysis in the spleen and liver (Risitano et al. 2009). Similar findings have not been observed in animals, likely because the toxicity only occurs in patients with PNH.
3.5. Cytokine-mediated Dysregulation of Erythropoiesis
Combination therapy of recombinant IFN-α (IFN-α2a or -2b, and peg-IFN-α2a or -2b) and ribavirin for hepatitis C results in anemia in approximately 10 to 30% of patients (Kowdley 2005). Although the anemia is largely due to ribavirin-induced nonimmune hemolysis, IFN suppresses erythroid progenitor cells and accounts for a decreased and delayed response of the bone marrow to decreased red cell mass (Kowdley 2005). Pegylated and nonpegylated IFN-α have also been associated with mild decreases in red cell counts that persist during the dosing period in nonhuman primate toxicity studies (PEG-INTRON [Peg-IFN-α2b] Toxicologists Review 2004; Pegasys [Peg-IFN-α2a] Preclinical Review 2009).
3.6. Anemia of Undetermined Mechanism
Rare cases of transient PRCA and hemolytic anemia have been reported with rituximab (chimeric mAb against CD20) monotherapy of lymphoproliferative disease in humans (Garratty and Petz 2007; Jourdan et al. 2003). These cases are suspected to be autoimmune; however, data supporting this hypothesis are limited (Gibbs et al. 2005). This type of anemia has not been reported in nonclinical studies.
4. Biotherapeutic-induced Neutropenia
Drug-induced neutropenia is an uncommon effect of biotherapeutics (Table 6). Attribution of neutropenia to a given biotherapeutic may be confounded by prior or concurrent administration of small molecule myelotoxicants or other biotherapeutics, particularly for cancer indications. The predominant complication of neutropenia (grade 3 or 4) is a higher risk of secondary infection, particularly when the bone marrow neutrophil reserve is depleted (Dinauer and Coates 2005).
Selected biotherapeutics associated with neutropenia.a

Note: IFN = interferon; IVIG = intravenous immunoglobulin. aDecreased neutrophils in nonclinical species may be within reference interval.
The pathogenesis of biotherapeutic-induced neutropenia is generally not known. Potential mechanisms include bone marrow suppression, direct cytotoxicity, drug-induced anti-neutrophil antibodies, and cytokine and complement pathway activation. The mechanism is undetermined for several biotherapeutics. Clinical grading categories for neutropenia are listed in Table 1.
4.1. Alemtuzumab
Administration of alemtuzumab, an anti-CD52 mAb, is associated with dose-related, grade 3 to 4 neutropenia in humans. Alemtuzumab-associated neutropenia occurs only after multiple doses and has an incidence of approximately 50% in some studies (Elter, Hallek, and Montillo 2011; Enblad et al. 2004; Gibbs et al. 2005; Lundin et al. 2003). Although the majority of cases of neutropenia resolve with removal of the drug, a small number of patients can develop pancytopenia refractory to granulocyte colony-stimulating factor (G-CSF) treatment (Elter, Hallek, and Montillo 2011; Enblad et al. 2004; Gibbs et al. 2005). Cynomolgus macaques administered alemtuzumab demonstrated reversible neutropenia only after repeated dosing in studies of 14 to 30 days in length (Campath-1H [alemtuzumab] Pharmacology/Toxicology Review 2001).
The pathogenesis of neutropenia related to alemtuzumab in humans and nonhuman primates is unknown. Neutropenia could be related to direct effects of alemtuzumab on marrow progenitor cells or altered hematopoiesis as a consequence of immune dysregulation. Alemtuzumab binds CD34-positive cells and a subset of myeloid and erythroid colony-forming cells (CFCs). However, alemtuzumab had no effect on in vitro CFC assays (Gilleece and Dexter 1993; Williams et al. 2000). Although neutrophils express a low level of CD52 (Ambrose, Morel, and Warrens 2009), direct binding and removal of circulating neutrophils does not explain the delayed neutropenia after several cycles of dosing.
4.2. TNF-α Inhibitors
Mild neutropenia has been documented in human patients predominately with first-generation biotherapeutics that block TNF-α in inflammatory disease. These inhibitors include adalimumab, infliximab, and etanercept. The second-generation anti-TNF-α biotherapeutics, certolizumab pegol (Cimzia [certolizumab pegol] label 2010) and golimumab have a very low frequency of neutropenia in comparison to the first-generation molecules (Storage, Agrawal, and Furst 2010). Neutropenia was not reported for first- or second-generation TNF-α inhibitors in nonclinical studies using cynomolgus macaques or chimpanzees.
Neutropenia with TNF-α inhibitors is generally low grade and nonprogressive (grade 1 or 2) and without infectious complications (Haroon, Daly, and Harney 2012; Hastings et al. 2010). Neutropenia occurs during the course of therapy, with a median onset 17 weeks after beginning treatment (range 3 days to 151 weeks). The combined incidence of neutropenia is 18% in patients with inflammatory arthritis given adalimumab, infliximab, or etanercept (Hastings et al. 2010). Risk factors for neutropenia with TNF-α inhibitors include a clinical history of neutropenia with other disease-modifying antirheumatic drugs and a low baseline neutrophil count. Whether neutropenia is a class effect of biotherapeutic-induced TNF-α inhibition is uncertain. The limited reports of neutropenia with several new generation inhibitors and recovery of neutrophil counts in some patients when given alternative first-generation TNF blockers (Hastings et al. 2010) suggest that patient factors, pharmacokinetics, and/or pharmocodynamics contribute to the effect.
The mechanism of anti-TNF-α neutropenia is unknown but is consistent with the peripheral removal of neutrophils rather than decreased production as bone marrow examination shows no abnormality in myelopoiesis (Wenham, Gadsby, and Deighton 2008). The membrane-bound form of TNF-α expressed by neutrophils binds first-generation TNF-α inhibitors and may contribute to peripheral removal (Mitoma et al. 2008). Higher expression of membrane-bound TNF-α on neutrophils in rheumatoid arthritis patients (Wright et al. 2011) may increase susceptibility to neutropenia. First-generation TNF-α inhibitors but not a second-generation molecule (certolizumab pegol) caused degranulation and apoptosis of human peripheral blood neutrophils in vitro (Nesbitt et al. 2007).
4.3. Rituximab
Rituximab (Rituxan) is a chimeric mAb against CD20. Rituximab is associated with late onset neutropenia, with an incidence of between 5.6 and 27.3% in lymphoma patients (Tesfa and Palmblad 2011). Late-onset neutropenia (LON) associated with rituximab is a recently recognized type of biotherapeutic-induced neutropenia generally defined as a self-limiting disorder that is diagnosed after 4 weeks postdose with a severity of at least grade 2 (<1.5 × 109 cells/L). Neutropenia of grades 3 and 4 (<1.0 × 109 cells/L; Tesfa and Palmblad 2011) is frequently observed with LON. Neutropenia did not occur in nonclinical studies up to 10 weeks in duration in cynomolgus macaques (Rituxin [rituximab] Preclinical and Clinical Pharmacology Review 1997).
Neutropenia occurs with rituximab alone or in combination with other drugs. The median time of onset for neutropenia is 56 to 175 days post-administration, with a median duration of 6 to 77 days (Tesfa and Palmblad 2011). The FcγRIIIa 158 V/F polymorphism is a predisposing factor for neutropenia, increasing the odds of rituximab-induced LON variously by 1.47-fold for VV/VF (Li et al. 2010) or 3-fold with each 158V allele (Cartron et al. 2002; Weng et al. 2010).
Bone marrow findings in patients with rituximab-associated neutropenia include granulocytic hypoplasia, a left shift of neutrophil precursors, and sometimes a maturation block at the myelocyte or metamyelocyte stage (Chaiwatanatorn et al. 2003; Fukuno et al. 2006; Stamatopoulos et al. 2008; Tesfa et al. 2008). Anti-neutrophil antibodies are uncommon (Tesfa and Palmblad 2011).
The mechanism of rituximab-associated LON is unknown. To date, no hypothesis accounts for the spectrum of changes including late onset, spontaneous resolution, and ineffective granulopoiesis in the bone marrow. The magnitude of B cell depletion and/or the rate of B cell recovery have been linked to LON (Dunleavy et al. 2005; Tesfa and Palmblad 2011). One theory is that neutropenia arises from impaired myelopoiesis due to altered growth factors and/or competition between cell lineages in the marrow niche during homeostatic restoration of B cells (Dunleavy et al. 2005; Terrier et al. 2007). Alternatively, Fas-mediated apoptosis of neutrophils by increased Fas associated with expansion of reactive granular T lymphocytes has been demonstrated by one group (Stamatopoulos et al. 2008) but has not been confirmed by other investigators (Fukuno et al. 2006; Terrier et al. 2007; Tesfa et al. 2008).
Second- and third-generation fully humanized anti-CD20 molecules are associated with grade 3 or 4 neutropenia during the dosing phase (discussed in section 4.4. on Monoclonal Antibodies with Enhanced Effector Function; Bello and Sotomayor 2007; Lemery et al. 2010; Tobinai et al. 2011; Wierda et al. 2010). However, late-onset neutropenia has not been reported to date in early clinical trials with these more potent molecules (Goldenberg, Morschhauser, and Wegener 2010; Lemery et al. 2010; Morschhauser et al. 2009, 2010).
4.4. Monoclonal Antibodies with Enhanced Effector Function
Monoclonal antibodies have been developed with Fc modifications to enhance effector function. The higher affinity of these antibodies to FcγRIIIa on macrophages and NK cells augments ADCC. Clinical cases of self-limiting neutropenia (grades 1–3) have been reported for 2 mAbs against CD20: ofatumumab and LY2469298 (Lemery et al. 2010; Tobinai et al. 2011; Wierda et al. 2010). In addition, neutropenia was reported in nonclinical monkey studies for RhuMAbv114 (Bello and Sotomayor 2007). These three mAbs target CD20, while neutropenia has not been reported with other mAbs with enhanced effector function. The mechanism for neutropenia associated with molecules with enhanced effector function is not known.
4.5. Direct Binding to Neutrophils—Intravenous Immunoglobulinu
IVIG has been associated with transient neutropenia occurring within days after administration to humans, but not with nonclinical species. IVIG-associated neutropenia is self-limiting and not complicated by secondary infection. Alloantibodies to neutrophil membrane components (e.g., Fas, sialic acid–binding immunoglobulin-like lectin [Siglec]-8, and Siglec-9 receptors) may be present in IVIG preparations and may cause direct neutrophil cytotoxicity (Baxley and Akhtari 2011). Pseudoleukopenia due to increased aggregation of neutrophils resulting in erroneously low cell counts has also been noted in blood of patients treated with IVIG (Zelster et al. 2000).
4.6. IFN-α-mediated Dysregulation of Neutrophil Production
IFN-α results in a dose-related, mild neutropenia in 8 to18% patients. Neutropenia occurs more frequently with peg-IFN-α2a or -2b compared to other IFN biotherapeutics (non-pegylated IFN-α2a or -2b; Kowdley 2005). Neutropenia may require dose reduction but is not associated with infection risk (Antonini et al. 2008; Kowdley 2005; Soza et al. 2002). The nadir of neutropenia mostly occurs within 4 weeks of dosing for peg-IFNα (Durante-Mangoni et al. 2006). Suppression of myeloid progenitor cells by IFN-α, as suggested by in vitro assays, likely accounts for the neutropenia (Broxmeyer et al. 1983). Inappropriately low levels of endogenous G-CSF have also been demonstrated in patients that have developed IFN-α-associated neutropenia (Durante-Mangoni et al. 2006). Pegylated and nonpegylated IFN-α-2b have also been associated with transient decreases in neutrophils in multidose toxicity studies in nonhuman primate studies. In nonhuman primates, the recovery of neutrophil counts during the dosing phase corresponded to the appearance of neutralizing antibodies which may have altered the toxicity profile (Peg- IFNα-2b [PEG-INTRON] Toxicologists Review, 2004).
4.7. Neutropenia of Undetermined Mechanism
Bevacizumab (Avastin) and trastuzumab (Herceptin) are humanized mAbs against VEGF and Her2/neu, respectively. These targets are not known to be expressed by the neutrophil lineage; however, both of these biotherapeutics have been associated with a low-incidence neutropenia in humans ranging from grade 1 to grade 4 through unknown mechanisms (Schutz et al. 2011). There have been no reports of neutropenia associated with bevacizumab and trastuzumab in nonclinical studies.
5. Biotherapeutics Causing Multiple Cytopenias as Secondary Effects
In contrast to the biotherapeutic-associated cytopenia discussed in previous sections, some biotherapeutics can cause primary toxicities, including hemophagocytosis, infusion reactions, and TMA. These primary toxicities can result in downstream effects on single or multiple blood cell types, which are “innocent bystanders” in the process (Table 7). These cytopenias result from destruction, consumption, altered trafficking and sequestration, and decreased production of blood cells.
5.1. Hemophagocytosis
Numerous biotherapeutics cause multiple cytopenias by excessive hemophagocytosis; the effects of these biotherapeutics are similar to the naturally occurring hemophagocytic syndrome (HPS). HPS is a group of related diseases in animals and humans characterized by phagocytosis of circulating blood cells by the monocyte–macrophage system, resulting in multiple cytopenias. Congenital or acquired HPS is usually associated with dysregulation of lymphocytes resulting in chronic stimulation of CD8+ T cells and excessive production of IFN-γ from T cells and macrophages, such as TNF-α, IL-1, IL-6, and IFN-γ (Janka 2007). Acquired HPS secondary to infections, neoplasia, and drugs has been reported in rats, dogs, rhesus macaques, and humans (Cotroneo, Colby, and Bergin 2011; Ide et al. 2009; Stromberg et al. 1983; Weiss 2007). Biotherapeutics causing hemophagocytosis include interleukins, hematopoietic growth factors, and mAbs, and are listed in Table 8.
Selected biotherapeutics associated with multiple cytopenias.

Note: IFN = interferon.
Selected biotherapeutics causing hemophagocytosis.

Note: rh = recombinant human; rm = recombinant murine; rc = recombinant canine.
5.1.1. Cytokines and Hematopoietic Growth Factors
Administration of cytokines and hematopoietic growth factors have been associated with hemophagocytosis resulting in moderate to severe thrombocytopenia and decreased red cell mass (Abrams et al. 2003; Baker and Levin 1998). In general, nonclinical results have predicted hemophagocytosis associated with recombinant cytokines and growth factors in patients (Table 8). The underlying cause of single or multiple cytopenias is likely activation of monocyte/macrophages in the spleen and/or liver, resulting in phagocytosis of platelets and red blood cells (Abrams et al. 2003; Baker and Levin 1998).
5.1.2. mAbY.1
Monoclonal antibody Y.1 (mAbY.1) is a fully human IgG2 mAb biotherapeutic against a human cell surface membrane-bound protein. In nonclinical studies, mAbY.1 caused dose-related profound thrombocytopenia (nadir ∼3,000 platelets/ul) with mild to marked decreases in red cell mass (Everds et al. 2013). Other mAbs sharing the same Fc framework and similar biological activity against the intended target did not have the same hematotoxicity in vitro or in vivo.
mAbY.1 directly or indirectly activates splenic macrophages, leading to phagocytosis of circulating platelets and red blood cells in the spleen. mAbY.1 does not bind to cynomolgus peripheral blood or bone marrow cells in vitro, as assessed by flow cytometry. In vitro, mAbY.1 induces phagocytosis of platelets by cynomolgus but not human peripheral blood monocytes. Modification of the Fc portion of mAbY.1 attenuates the in vivo and eliminates the in vitro hematologic responses. These data suggest that hemophagocytosis caused by mAbY.1 in cynomolgus monkeys occurs through a mechanism involving both the Fc and complementarity determining region (CDR) of the mAb.
5.1.3. Anti-EGFL7
Anti-EGFL7 is a humanized mAb that cross-reacts with human, cynomolgus monkey, and rat EGFL7, a protein secreted by endothelium (Couch et al. 2010). Dose-related decreases in mean platelet counts of approximately 25 to 33% occurred in rats but not in cynomolgus monkeys. By quantitative polymerase chain reaction (qPCR), EGFL7 expression was approximately 3- to 5-fold greater in the spleen of rats compared to humans and cynomolgus monkeys. Splenic macrophages of rats dosed with anti-EGFL7 were hypertrophied and contained platelets in phagolysosomes, suggesting that anti-EGFL7 administration to rats resulted in platelet phagocytosis by splenic red pulp macrophages with a concomitant decrease in circulating platelets. Clinical data have not been reported for this biotherapeutic.
5.2. Infusion Reactions
Infusion reactions are commonly reported after biotherapeutic therapy in both nonclinical and clinical studies. The term infusion reaction is used to describe a spectrum of clinical changes occurring in subjects during or shortly after intravenous dosing (Kang and Saif 2007). These signs can range from mild to fatal and include fever, chills, myalgia, hypotension, nausea, vomiting, headache, urticaria, dyspnea, bronchospasm, and cardiovascular collapse (Chung 2008; Dillman 1999; Lenz 2007; Vogel 2010). Although infusion reactions are sometimes incorrectly referred to as hypersensitivity reactions, true hypersensitivity (e.g., types I–IV of the Coombs-Gell classification) accounts for only a portion of infusion reactions.
Biotherapeutics that are most often associated with infusion reactions in humans include rituximab, (anti-CD20), muromonab (anti-CD3), infliximab (anti-TNF-α), and alemtuzumab (anti-CD52; Carney and Ollom 2008; Chung et al. 2008; Keystone 2001; Maggi, Vultaggio, and Matucci 2011). Underlying causes of infusion reactions in response to biotherapeutics are heterogeneous and include complement activation, cytokine release, and hypersensitivity reactions (Daguet and Watier 2011; Maggi, Vultaggio, and Matucci 2011; Vogel 2010). Activation of complement or a cytokine cascade occurs after a single dose. In contrast, most but not all hypersensitivity (immunoglobulin-mediated) infusion reactions occur after multiple doses and are due to drug-specific or cross-reacting IgG and/or IgE. Complement can be secondarily activated after cytokine release and by anti-drug IgG/drug complexes (Tawara et al. 2008).
In nonclinical studies, biotherapeutics are often administered via an intravenous bolus injection rather than a slow infusion, which may increase the incidence or severity of infusion reactions. In contrast, biotherapeutics are often administered to patients at a slower rate in an intravenous infusion. Most infusion reactions in patients are managed by decreasing the rate of administration of drug and/or by supportive therapy such as administration of antihistamines and corticosteroids (Calogiuri et al. 2008; Carney and Ollom 2008; Chung 2008; Kang and Saif 2007; Lenz 2007; Mayer and Young 2006; Patel and Goldberg 2006).
Infusion reactions in nonclinical studies due to complement activation are generally predictive for humans due to the conserved nature of the complement system, but infusion reactions due to ADAs are not predictive for humans because immunogenicity of a human protein in animals does not predict human immunogenicity (Bugelski and Treacy 2004; Dixit and Boelsterli 2007; ICH S6 1997; Ponce et al. 2009).
Clinical signs associated with infusion reactions after biotherapeutic therapy in animals and humans do not differentiate the underlying cause of the reaction. Due to the common clinicopathologic findings, biotherapeutics causing infusion reactions are described by mechanism in this review. Whether or not cytopenias occur with a given infusion reaction depends on the underlying cause.
5.2.1. Complement Activation
Several classes of biotherapeutics have been shown to exert unexpected hematologic effects due to activation of complement, including oligonucleotides, aptamers, and liposomes (Szebeni 2005). These direct complement-mediated hypersensitivity reactions have been termed complement-activation-related pseudoallergy (CARPA) due to similarities of their clinical signs to IgE and mast cell–mediated anaphylaxis. CARPA is distinguished from anaphylaxis by the following characteristics: (1) CARPA occurs after the first exposure to a test article; (2) subjects become refractory to the complement-mediated effects upon subsequent doses due to complement depletion; (3) effects do not depend on antigen-specific IgE; and (4) the incidence is higher (5–45%) for complement-mediated events compared to anaphylaxis (<2%; Szebeni et al. 2007).
Hematologic effects of CARPA vary among species and on the therapeutic agent. Rats are more resistant to complement hypersensitivity reactions from liposomes compared to pigs, dogs, and humans (Szebeni et al. 2007). Rats exhibit thrombocytopenia and hemoconcentration; pigs primarily exhibit cardiopulmonary toxicities rather than hematologic changes; and dogs exhibit thrombocytopenia and leukopenia followed by leukocytosis (cell type unpublished). Complement activation resulting in a biphasic leukocyte response has been described in macaques administered selected oligonucleotides (Galbraith et al. 1994; Henry et al. 2002). Complement activation in macaque monkeys is characterized by neutropenia followed rapidly by neutrophilia, with or without thrombocytopenia (Galbraith et al. 1994).
In humans, complement activation is responsible for thrombocytopenia associated with infusion of TNF-α and IFN-γ for up to 7 days (Nurnberger et al. 1994). The thrombocytopenia can be moderate or profound depending on the duration of the infusion. Thrombocytopenia is considered secondary to complement activation, with possible contribution from endothelial activation (Michelmann et al. 1997). Complement activation is also a factor in cytopenias associated with rituximab (see 5.2.2. Cytokine Release below).
5.2.2. Cytokine Release
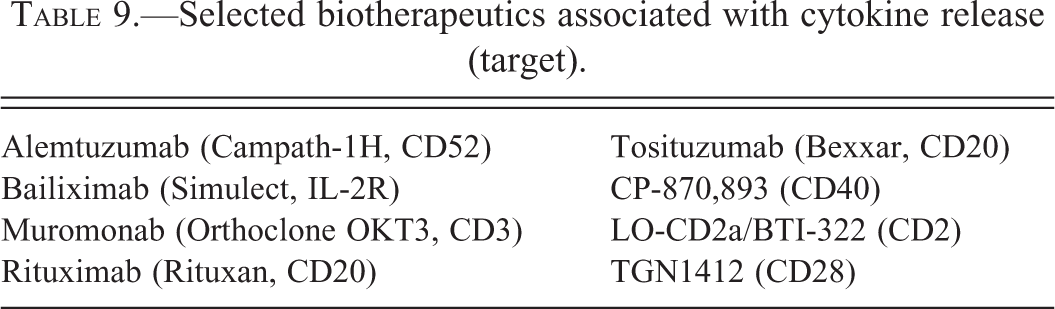
Several mAbs, including those listed in Table 9, have been associated with altered blood cell counts due to cytokine release (Bugelski et al. 2009). Administration of these drugs can be associated with immediate release of TNF-α and IFN-γ, followed by later release of IL-6. The underlying mechanism for cytokine release is variable among mAbs.
Selected biotherapeutics associated with cytokine release (target).
Cytokine release may occur only after the first dose for drugs that bind to and deplete target immune cells (e.g., muromonab, rituxumab, and alemtuzumab; Gribble et al. 2007). Hematologic effects associated with cytokine release syndromes have been described for TGN1412 and rituximab and include lymphopenia, monocytopenia, and thrombocytopenia. Reported hematologic effects vary depending on the degree of cytokine release and the time at which samples are collected.
Patients treated with TGN1412 had lymphopenia and monocytopenia at 24 hours postdose (Suntharalingam et al. 2006). Neutrophil counts were unaffected, but neutrophil morphological changes were present, including Döhle bodies, toxic granulation, and pseudo Pelger-Huët morphologic changes. At later time points, patients had moderate thrombocytopenia attributable to disseminated intravascular coagulation.
Patients treated with rituximab can develop acute reversible thrombocytopenia due to cytokine release and complement activation (Ram et al. 2009). Rituximab-associated thrombocytopenia is associated with clinical symptoms of cytokine release syndrome in 82% of thrombocytopenic patients. Rituximab-associated thrombocytopenia begins within hours of dosing, reaches a nadir within 1 day after dosing and resolves spontaneously (Dhand and Bahrain 2008).
In some instances, nonclinical in vivo studies may not predict clinical cytokine release (Bugelski et al. 2009). Recently, several groups have published on in vitro assays that may offer predictivity for in vivo cytokine release (Dhir et al. 2012; Findlay et al. 2011).
5.2.3. Hypersensitivity Reactions
Hypersensitivity (Coombs type I or III) reactions associated with biotherapeutic administration generally occur after multiple doses because they require the de novo development of antibodies to the drug (Calogiuri et al. 2008). Some hypersensitivity reactions that occur after a single dose of a biotherapeutic are thought to be due to preexisting or natural antibodies that cross-react with the therapeutic molecule. For oncologic biotherapeutics, hypersensitivity reactions occur in approximately 10 to 40% of patients (Lenz 2007). Although hypersensitivity reactions can be observed with any biotherapeutic, they are more commonly reported in association with infliximab, rituximab, and alemtuzumab (Hellerstedt and Ahmed 2003; Krishnan and Hsu 2004).
ADA-related hypersensitivity reactions in nonclinical species and humans can be driven by IgG (type III hypersensitivity)- or IgE (type I)-mediated hypersensitivity (Cheifetz et al. 2003; Maggi, Vultaggio, and Matucci 2011; Pichler 2006; Vogel 2010). Documented antidrug IgE resulting in anaphylaxis has been observed with numerous biotherapeutics in cynomolgus monkeys and humans (Abramowicz, Crusiaux, and Goldman 1992; Baudouin et al. 2003; Bremmer et al. 2009; Chung et al. 2008; Cox 2009; Dietrich et al. 2001; O'Neil et al. 2007; Werner, personal communication, 2012). Hypersensitivity due to antidrug IgE is considered to be less common than that due to IgG (Calogiuri et al. 2008). The lack of hematologic effects is characteristic of type I IgE-mediated hypersensitivity reactions and is in contrast to type III hypersensitivity reactions that are often characterized by cytopenias.
5.2.3.1. IgG-mediated hypersensitivity (type III, immune complex)
Type III IgG-mediated hypersensitivity can occur in nonclinical studies and in human patients when subjects develop antidrug IgG antibodies after administration of a biotherapeutic (Cheifetz et al. 2003; Proctor et al. 2004). Because human mAbs are more immunogenic in nonhuman primates, IgG-mediated hypersensitivity is likely more common in nonhuman primates. In the authors’ experience, marked hematologic effects in nonhuman primates due to IgG-mediated hypersensitivity are generally observed only after intravenous administration. These hematologic effects of type III hypersensitivity reactions after intravenous dosing of a biotherapeutic are generally transient due to the rapid removal (between 11 and 60 min) of immune complexes from circulation (Davies et al. 1990; van der Laken et al. 2007).
Clinical signs of hypersensitivity after intravenous dosing usually occur immediately during or after dosing, or within a few days of dosing. Symptoms are generally transient and most animals and patients recover; however, death can occur in severe reactions (Cheifetz et al. 2003; Ljungstrom et al. 1988; Todd and Helfgott 2007).
In nonhuman primates, the primary hematologic effect of IgG-mediated hypersensitivity after intravenous dosing of an mAb to a sensitized subject is a decrease in platelet counts due to platelet activation. Additional effects include decreased neutrophils and monocytes, with no change in lymphocytes; these leukocyte changes are mediated by complement (Birmingham et al. 1999; Davies et al. 1990; Smedegard, Revenas, and Saldeen 1980). The decreases in platelets are independent of complement in macaques, in contrast to rodent studies that implicate complement as the cause of decreases in both neutrophils and platelets (Birmingham et al. 1999; Kinsell et al. 1941; Kopeloff and Kopeloff 1941). Immune complexes cause thrombocytopenia in primates by cross-linking of FcγRIIa on the platelet surface, resulting in activation. Platelet activation can result in pulmonary thromboemboli, complement activation, and release of vasoactive amines that cause pulmonary venoconstriction (Del Conde et al. 2005; Hedin and Smedegard 1979; Radegran, Drugge, and Olsson 1974; Radegran and McAslan 1972; Revenas, Smedegard, and Saldeen 1980; Smedegard, Revenas, and Arfors 1979).
Hematologic effects of type III hypersensitivity are poorly documented in humans (Arimura et al. 2004; Calogiuri et al. 2008; Pichler 2006). Humans with IgG-mediated hypersensitivity secondary to administration of high-molecular-weight dextran had severe clinical signs including cardiac arrest and prominent thrombocytopenia (Ljungstrom et al. 1988). The severity of acute infusion reactions correlated with anti-dextran IgG titers from samples obtained prior to dosing (Hedin and Richter 1982). The classical pathway of complement was also activated in these patients.
Adalimumab and infliximab are associated with type III hypersensitivity and infusion reactions. A higher incidence of antidrug IgG predisposes patients to thromboembolic events (adalimumab) and infusion reactions (infliximab; Baert et al. 2003; Korswagen et al. 2011; Wolbink et al. 2006). Similarly, IgA-deficient humans administered IVIG or other blood products may have similar hypersensitivity reactions (Leikola et al. 1973). To the authors’ knowledge, platelet counts have not been reported for these subjects.
5.3. Thrombotic Microangiopathy
TMA is a group of familial or acquired disorders characterized by thrombosis in microvasculature with secondary hemolysis (decreases in red cell count, increased reticulocyte count, and red cell fragments on blood smear examination) and thrombocytopenia. TMA includes two major syndromes, TTP and hemolytic uremic syndrome (HUS), and is sometimes associated with deficient von Willebrand factor–cleaving protease (ADAMTS13) and decreased complement regulatory factor activity, respectively (Blake-Haskins, Lechleider, and Kreitman 2011). Conditions resembling TTP and HUS have been observed with several biotherapeutics; however, the underlying mechanism is undetermined.
5.3.1. Bevacizumab
Bevacizumab (a humanized mAb to VEGF-A) has been associated with symptoms of TTP, with findings of microangiopathic hemolytic anemia, thrombocytopenia, hypertension, and proteinuria in humans but not in nonclinical species (Eremina et al. 2008). In most cases, thrombosis has been confined to the kidney, likely due to the importance of VEGF in maintaining glomerular endothelial integrity (Eremina et al. 2008). Combination therapy of bevacizumab and sunitinib (a small molecule receptor tyrosine kinase inhibitor that inhibits VEGF) was associated with TMA characterized by proteinuria and neurologic signs in 5 of 25 renal cell carcinoma patients (Feldman et al. 2009).
5.3.2. IFN-α and Muromonab-CD3
IFN-α therapy and muromonab-CD3 have induced TTP in rare cases in humans (Pisoni, Ruggenenti, and Remuzzi 2001) and has not been described in nonclinical studies. A deficiency in ADAMTS13 was demonstrated in some cases of TTP associated with peg-IFNα-2a, suggesting a similar pathogenesis to the naturally occurring condition (Kitano et al. 2006). In addition, mild reversible symptoms resembling HUS have been reported at incidences reaching 10% in early clinical trials with the immunotoxins DAB486IL-2, CAT3888, Moxetumomab pasudotox, and Combotox (Blake-Haskins, Lechleider, and Kreitman 2011).
5.4. Multiple Cytopenias Due to Undetermined Mechanisms
5.4.1. Alemtuzumab
Pancytopenia has been reported in a small number of patients given alemtuzumab, generally after 5 weeks of treatment. Bone marrow findings suggesting effects of alemtuzumab on hematopoiesis and blood cell survival have included hypoplasia, trilineage myelodysplasia, and/or hemophagocytosis (Enblad et al. 2004; Gibbs et al. 2005). Pancytopenia in these patients could be due to direct effects of the drug on hematopoiesis (e.g., alemtuzumab-mediated cytotoxicity or selection of CD52-negative clones) or indirect effects of immune dysregulation leading to hemophagocytosis and decreased hematopoiesis. The presence of intercurrent disease in these patients (e.g., Epstein–Barr virus or histoplasmosis) and underlying T-cell lymphoproliferative disease obfuscates the relationship of alemtuzumab with cytopenias (Gibbs et al. 2005). Pancytopenia was not reported in nonclinical studies.
5.4.2. TNF-α Inhibitors
Rare cases of pancytopenia due to bone marrow aplasia or hemophagocytosis have been reported for the first-generation TNF-α inhibitors etanercept and infliximab (Araki et al. 2011; Desai and Furst 2006; Francolla, Altman, and Sylvester 2008; Kuruvilla et al. 2003; Vidal, Fontova, and Richart 2003). Pancytopenia (not associated with aplasia) has also been noted with adalimumab (Desai and Furst 2006). Determining whether administration of TNF-α is causal for cytopenias in these cases is difficult; other potential causes include the underlying disease, comedications, or secondary infection or autoimmune disease. Pancytopenia was not reported in nonclinical studies.
6. Summary and Conclusion
Biotherapeutics may cause hematotoxicity as a result of cellular activation, cytotoxicity, drug-dependent and independent immune responses, and sequelae from initiating cytokine and complement cascades.
Circulating blood cells are particularly susceptible to biotherapeutic-related toxicity for several reasons. Biotherapeutics administered intravenously are able to exert systemic effects immediately. When administered intravenously or subcutaneously, biotherapeutics often have long systemic exposures, resulting in prolonged periods of activity and increased potential for effects on blood cells. Many biotherapeutics target soluble or cell-surface proteins of the immune system that have a complex interactive relationship with bystander (nontargeted) blood cells. Blood cells, especially neutrophils, monocytes, and platelets, are primed for activity and can be activated by interaction of biotherapeutics with cell surface receptors. Additionally, processes resulting in infusion reactions can impact peripheral blood cells. Infusion reactions are the result of different mechanistic pathways that produce similar clinical signs and include hypersensitivity reactions but also downstream effects of complement activation and cytokine release.
Nonclinical animal studies have variable predictability for biotherapeutic-induced hematotoxicity. This review likely underrepresents translatability of nonclinical hematoxoticity to humans because nonclinical hematotoxicity may terminate the development of some biotherapeutics prior to human dosing, and the authors had access only to published data. Neutralizing antibodies to biotherapeutics in nonclinical studies may also affect the incidence, severity, and reversal of hematotoxicity reported in these studies. The concordance of negative findings between nonclinical and clinical studies is generally recognized as high for biotherapeutics; this explains the predominance of rare and idiosyncratic clinical hematotoxicity described in this review.
Of the limited number of biotherapeutics reviewed in this article, nonclinical studies predicted clinical hematotoxicity for hemophagocytosis resulting from recombinant cytokines and growth factors and thrombocytopenia caused by anti-CD40L mAbs. Nonclinical studies poorly predicted species-specific, immune-mediated, and low-incidence hematotoxicity. The lack of predictivity of nonclinical studies for low-incidence human hematotoxicity may be due in part to the small numbers of animals tested compared to the much larger number of humans that are administered drug during clinical trials. Several instances of high-incidence hematologic effects in nonclinical animal studies have been demonstrated to be species-specific upon in vitro mechanistic investigations or in vivo human dosing.
Unintended hematological effects of biotherapeutics, like small molecules, are sometimes not recognized until the post-marketing phase. Establishing that a given biotherapeutic causes hematotoxicity requires sufficient cases to distinguish drug-related hematological effects from confounding effects of primary and secondary diseases and prior and concurrent medications. Despite the potential for unexpected hematologic toxicity, the risk–benefit profile of the majority of these biotherapeutics is favorable. Potential hematologic effects are readily monitorable and managed by dose modification, drug withdrawal, and/or therapeutic intervention.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.