
Abstract
There is increasing evidence suggesting links between exposure to environmental toxins and susceptibility to type 2 diabetes mellitus (DM). In this review, we summarize the experimental evidence to support this association that has been noted in many epidemiologic studies. Inflammation in response to particulate matter (PM2.5) exposure in air pollution represents a common mechanism that may interact with other pro-inflammatory influences in diet and life style to modulate susceptibility to cardiometabolic diseases. The role of innate immune cytokines released from macrophages in the lung is well known. In addition, chemokine triggers in response to air-pollution exposure may mediate a cellular response from the bone marrow/spleen through toll-like receptors (TLRs) and Nucleotide Oligomerization Domain receptors (NLRs) pathways to mediate inflammatory response in organs. Emerging data also seem to support a role for PM2.5 exposure in endoplasmic reticulum stress-induced apoptosis and in brown adipose tissue dysfunction. Decreased expression of UCP1 in brown adipose tissue may account for reduced thermogenesis providing another link between PM2.5 and insulin resistance. The implications of an experimental link between air-pollution exposure and type 2 DM are profound as air pollution is a pervasive risk factor throughout the world and even modest alleviation in exposure may provide substantial public health benefits.
Keywords
Introduction
Chronic cardiometabolic diseases such as type 2 diabetes mellitus (DM) have become an uncontrolled global epidemic and a burgeoning cause of morbidity and mortality (http://www.idf.org/fact-sheets/diabetes-cvd). As a consequence of rapid increase in combustion from fossil fuels for power generation and transportation, air pollution has become a major health risk factor in many developing countries, in addition to some regions in the developed countries. Air pollutant levels in many parts of the world often exceed current regulatory standards. Given the worldwide burden of air pollution effects, its continuous and omnipresent nature, even small adverse health associations for individuals represent an enormous public health issue that deserves broad changes in public health policy and human behavior (Brook et al. 2010). This review summarizes recent epidemiological and experimental evidences on how air pollution may represent an under risk factor for the development of insulin resistance (IR) and type 2 DM. An understanding of the mechanisms underpinning this link may provide opportunities for the reduction of current air pollutant levels and may propose a cost-effective intervention in diabetes control.
Epidemiology of Air Pollution–associated Cardiometabolic Disease
Several epidemiological studies have demonstrated a positive association between particulate matter or traffic-related air pollutants and type 2 DM. Using the Ontario Health Insurance plan database, Brook et al. (2008) conducted one of the first cohort studies demonstrating an association between type 2 DM prevalence and air pollution in Ontario, Canada. Originally, the Canadian cohort was assembled for the purposes of studying the health impact of air pollution with cases of diabetes ascertained clinically. The exposure assessment was based on field measurements and land use regression models capable of predicting fine scale variation of NO2 levels, as a surrogate for traffic related air pollution. Logistic regression models adjusted for age, body mass index (BMI), and income revealed a positive relationship between NO2 exposure and DM prevalence (odds ratio [OR] 1.04; 95% confidence interval [CI]: [1.00, 1.08]) in women but not in men. In a cross-sectional ecological study by Pearson et al., (2010), the relationship between PM2.5 levels and diabetes prevalence in the United States was assessed by multivariate regression models using data obtained from the national diabetes surveillance system at the centers of disease control (CDC). Data for the annual mean PM2.5 were obtained for each county in the United States from the Environmental Protection Agency (EPA). Multiple covariates were examined including BMI > 30 kg/m2, physical activity, population, fast-food restaurant density, latitude, and socioeconomic data. The results revealed a significant association between PM2.5 levels and diabetes prevalence after adjustment for multiple covariants including exclusion of minority populations in the South, which have a high prevalence of DM. These results suggest 10,000 additional cases of DM/10 µg/m3 increase in PM2.5 (overall increase in prevalence of 1%/10 µg/m3). One important aspect of the analysis was that even when restricted to counties with PM2.5 < 15 µg/m3 (annual U.S. EPA limit) the findings remained significant suggesting that the relationship seems to extend to pollution levels considered “within national standards.”
Recently, the incidence of new DM in relation to air pollutants was evaluated in a prospective manner, adding additional support for the relationship between air-pollution levels and diabetes. The Danish Diet Cancer and Health Cohort consisted of 57,053 subjects, of whom 51,818 were eligible and followed up for 9.7 years. NO2 levels were used as a proxy for air-pollution exposure. The overall prevalence of DM in this cohort was low (5.5%). Air-pollution indices were associated with DM on multivariate models adjusted for covariants including [OR for NO2 levels 1.04 (1.01 to 1.04)] (Andersen et al. 2012). Another cohort study in Europe investigated 1,775 women aged 54 to 55 years without diabetes at enrollment in the highly industrialized Ruhr Valley of Germany. The hazard for diabetes was increased by 15 to 42% per interquartile range of PM10 or traffic-related exposure assessed using NO2 levels over 16 years. The associations persisted when different spatial scales were used to assess exposure and remained robust after adjusting for age, BMI, socioeconomic status, and exposure to several nontraffic-related sources of air pollution (Kramer et al. 2010). Sensitivity analyses indicated that women with high C3c blood levels (a complement fragment) were more susceptible for PM-related excess risk of diabetes than were women with low C3c levels. Interesting results from the Black Woman’s Health Survey have also shown an association between new onset DM and long-term exposure to NO2 (Incidence Rate Ratios: 1.25; 95%CI [1.07, 1.46]), a metric of traffic-related pollution. This relationship was shown among African American women living within the greater Los Angeles area (Coogan et al. 2012). On the other hand, the findings from the pooled analyses of the Nurses’ Health Study and the Health Professionals’ Follow-Up were less compelling (Puett et al. 2011). Close proximity of residence location to roadways and some metrics of traffic pollution exposures were modestly associated with DM incidence; however, multiple other exposure metrics were not significant.
Together, these epidemiological findings support the association between air pollution, in particular traffic-related sources, and DM. Nonetheless, not all aspects of this relationship have been consistently reported nor are they fully elucidated at this time. The varying associations noted between studies may relate to numerous differences. These include the population characteristics, risk factors, individual susceptibilities, robustness of the cohort data and the absolute prevalence/incidence rates of DM, technical aspects of the exposure assessment methodologies, pollution types/sources, and the degree and duration of air-pollution exposures. The sex-specific differences seen in some of these studies may relate to true differences in biologic susceptibility, a finding mirrored by observations in the Women’s Health Study that also demonstrated a greater susceptibility of obese women to air-pollution-mediated cardiovascular events (Miller et al. 2007). On the other hand, it is also possible that the sex predilection may relate to exposure assessment error particularly in males who tend to be more mobile compared to females. Notably, there are only limited studies on the association between air pollution and DM (or metrics of insulin sensitivity) in populations that suffer from exposure to very high levels of air pollutants. We are aware of only 2 relevant publications. In a study of 374 children in several Iranian cities, independent associations between recent 7-day exposure to particulate air pollution and plasma markers of inflammation, oxidative stress and insulin resistance were noted, which remained significant after adjustment for age, gender, BMI, waist circumference, healthy eating index, and physical activity level (Kelishadi et al. 2009). PM10 levels were extremely high in these cities, averaging roughly 150 µg/m3. In another study conducted in Taiwan, year-long fine particle exposure (mean level ≅ 35 µg/m3) was associated with elevations in HbA1c among 1,023 elderly individuals. Though these studies do not report the prevalence or incidence of overt DM, they do suggest that insulin sensitivity (the pathological hallmark underlying cardiometabolic disease) is worsened by exposure to particulate air pollution at very high concentrations (Chuang et al. 2011). In this context, the dose–response relationship and potential threshold concentrations for health responses (e.g., level above which the cardiometabolic effects are saturated and no longer worsened [high end] or below which they are no longer apparent [low end]) require clarification in future studies.
Potential Biological Mechanisms of Air-pollution-mediated Type 2 DM
A potential link between signals perceived in the lung such as air pollution and susceptibility to chronic metabolic disease may occur through a multitude of mechanisms reviewed in the following paragraphs. Some of these mechanisms originate in the lung via direct release of inflammatory cytokines, generation of secondary mediators in the lung or via direct translocation of particles. Secondary innate and adaptive immune responses have also been described. Autonomic neural mechanisms may also underlie a number of responses. The next few paragraphs will attempt to review this information.
Pulmonary and Systemic Inflammation
In general, PM is considered to represent a prototypical inflammatory trigger with studies demonstrating a range of abnormalities in the lung that include cellular response including innate and adaptive immune cells and release of inflammatory cytokines. There may also be significant synergistic effects with other cells such as bronchial epithelial cells in production of such cytokines. Elevations in systemic and pulmonary levels of IL-6 and TNF-α have been observed in experimental animal models following PM exposure, typically coincident with pulmonary inflammation (Becher et al. 2007; Boland et al. 1999; Fujii et al. 2002; Quay et al. 1998; Shukla et al. 2000; Tamagawa et al. 2008; Tornqvist et al. 2007; van Eeden et al. 2001). Moreover, there is at least some evidence that the degree of pulmonary inflammation correlates with the elevation of systemic cytokines and systemic vascular dysfunction (Tamagawa et al. 2008). Several controlled human exposure studies have measured systemic inflammatory cytokines following exposure to PM, and while some studies have found elevations, others have not (Carlsten et al. 2007; Mills et al. 2007, 2005; Tornqvist et al. 2007). The lack of an association between the vast majority of acute exposure studies and inflammatory markers, however, may not preclude effects with chronic exposure nor does it preclude an effect on other cytokine pathways that have not been investigated.
Two studies with controlled exposures have shown increase in white cell indices following exposure. In one study, increased peripheral basophils in healthy older adults 4-hr following a 2-hr exposure to PM2.5 were noted (Gong et al. 2004). In another study, increased white blood cell counts in healthy young adults 12-hr following a 2-hr exposure to PM2.5 concentrated ambient particles were noted (Ghio et al. 2003). Frampton et al. reported decreases in blood monocytes, basophils, and eosinophils following exposure to ultrafine carbon (10 to 50 µg/m3) among exercising asthmatics and healthy adults (Frampton et al. 2006). Particle exposure also reduced expression of adhesion molecules CD54 and CD18 on monocytes. These results may be interpreted as increased sequestration of these cells in tissue compartments such as the lung or vasculature where there may be selective expression of the corresponding receptors for these ligands (Yatera et al. 2008). Other recent human clinical studies have found no association between peripheral blood cell counts and exposure to fine or ultrafine particles such as zinc oxide (Beckett et al. 2005), ultrafine carbon (Routledge et al. 2006), or diesel exhaust (Mills et al. 2007, 2005; Tornqvist et al. 2007). In the only study to date to examine the effects on specific subsets of peripheral cells, Peretz et al. (2007) evaluated gene expression using an expression array in monocytes following 4 hr of exposure to diesel exhaust. Ten genes involved in the inflammatory response were modulated in response to PM2.5 exposure (8 upregulated and 2 downregulated). These findings raise the possibility that functional changes in inflammatory cells may occur without discernible changes in counts of these cells in the peripheral circulation (Peretz et al. 2007). Moreover, with the expanding complexity of inflammatory subsets of cells better and more sensitive flow-sorting methodologies may allow discernment of effects in specific populations of cells (Charo 2007; Geissmann et al. 2008).
PM2.5-mediated Alterations in Glucose Homeostasis
PM2.5-mediated elevations in blood glucose levels have been shown in mice fed both on normal diet (ND; Figure 1A) and on high-fat diet (HFD) in our group (Sun et al. 2009; Xu et al. 2011). Defective insulin signaling in tissues such as the liver is fundamental to the pathogenesis of IR/DM. In keeping with this, we noted decreased tyrosine phosphorylation in the liver with PM2.5 exposure without changes in insulin receptor substrate (IRS) levels. Alteration in IRS phosphorylation in insulin resistance has been shown previously to result in defective PI3K-Akt signaling and suppression of insulin stimulated glucose transporter translocation. We noted reduced Akt phosphorylation in the liver, skeletal muscle, white adipose tissue (Figure 1), and aorta at both baseline and in response to insulin stimulation, indicating development of insulin resistance in multi-organs. Our findings have been replicated by other investigators (Yan et al. 2011).
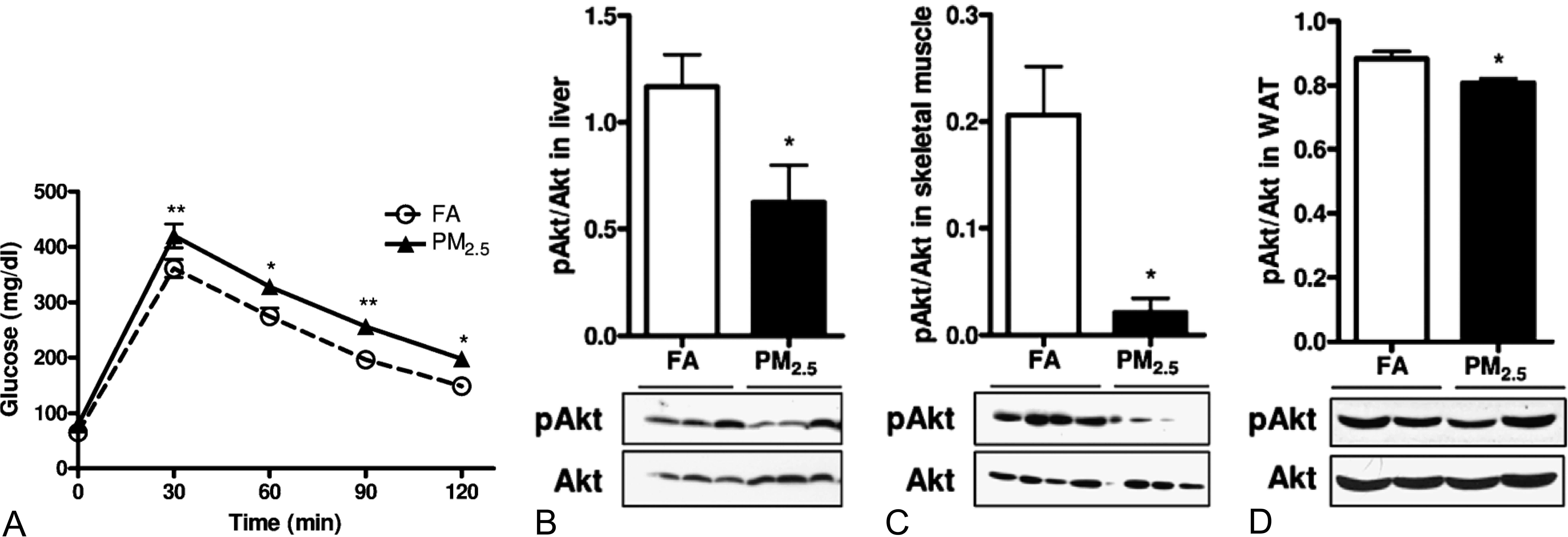
Effect of PM2.5 exposure on glucose homeostasis by intraperitoneal glucose tolerance test (A) and Western blotting showing phosphorylated (p-Akt at serine 473) and total Akt levels in the liver (B), soleus skeletal muscle (C), visceral WAT (D). *p < .05, **p < .01 versus FA group. Data were from C57BL/6 mice fed a normal diet and exposed to PM2.5 or filtered air for 10 months. (Modified with permission from Xu et al., Toxicol Sci, 2010.)
PM2.5-mediated Inflammation in Visceral Adipose Tissue
Type 2 DM in humans and animal models is associated with increased levels of recruitment and/or activation of innate immune cells in visceral adipose depots. PM2.5 exposure results in an increase in adipose tissue macrophages with a shift to a pro-inflammatory phenotype characterized by an increase in F4/80 macrophages in the visceral adipose and a pro-inflammatory “M1 phenotype” typified by TNF-α, IL-6 and a decrease in IL-10, MgI1 gene expression (Figure 2A and B; Sun et al. 2009). In order to further assess the ability of PM2.5 exposure in mediating infiltration of visceral adipose tissue, we assessed the effects of intra-tracheally delivered PM2.5 in a transgenic model of yellow-fluorescent protein expression restricted to monocytes (c-fmsYFP ). After rendering these mice insulin resistant with HFD, PM2.5 exposure resulted in a doubling in the number of endothelial adherent YFP+ cells in mesenteric fat with a 6-fold increase in monocytes within adipose (Figure 2C; Sun et al. 2009). Thus, PM2.5 facilitated migration and adhesion of YFP+ cells into visceral fat depots. In subsequent experiments, we investigated the effects of early (at age of 3 weeks) PM2.5 exposure on development of insulin resistance with mice that were exposed to both ND and HFD. Mice on ND exposed to PM2.5 showed significant elevations in glucose levels by an intraperitoneal glucose tolerance test (Figure 3A and B). HFD (regardless of FA or PM2.5 exposure) and PM2.5 exposure in ND-fed mice led to elevations in homeostasis model assessment index-insulin resistance (HOMA-IR; Figure 3C) and elevations in TNF-α compared with the FA in either ND- or HFD-fed mice (Figure 3D). PM2.5 exposure alone and HFD feeding significantly increased the total abdominal fat compared with FA-exposed mice fed ND. Both visceral and subcutaneous fat content were increased with PM2.5 exposure in the ND group (data not shown). Adipocyte size was increased in the PM2.5-exposed mice fed an ND in both visceral fat (FA, 2,137 ± 45 µm2; PM2.5, 2,698 ± 80 µm2; p < .01) and subcutaneous fat (FA, 1,039 ± 27 µm2; PM2.5, 1,355 ± 30 µm2; p < .05). The increase in adipocyte size, was however, extreme in the HFD group alone, not allowing any further changes due to PM2.5 exposure. These data suggest that PM2.5 exposure alone, in the presence of ND, may potentiate adiposity and exert pro-inflammatory effects. In light of the importance of NADPH oxidase in mediating the altered metabolic profile and insulin resistance, age-matched male p47phox−/− mice were exposed to PM2.5 or FA using the same exposure protocol as the wild-type C57BL/6 mice. HOMA-IR indexes from the PM2.5-exposed p47phox−/− mice were significantly attenuated and comparable to those of the FA-exposed mice fed an ND. Plasma inflammatory biomarkers in the p47phox−/− mice were similar to those in the wild-type C57BL/6 mice. Notably, the absence of a functional NADPH oxidase abrogated the previously noted difference in TNF-α in wild-type C57BL/6 with PM2.5 exposure (Figure 3E–H). In p47phox−/− mice, adipocyte size in the PM2.5-exposed mice fed an ND was similar to that in the FA-exposed mice on the same diet in both visceral and subcutaneous fat. PM2.5 exposure alone (normal chow diet) resulted in a heightened chemotactic ability of adipose tissue from PM2.5 exposed mice (Xu et al. 2010).
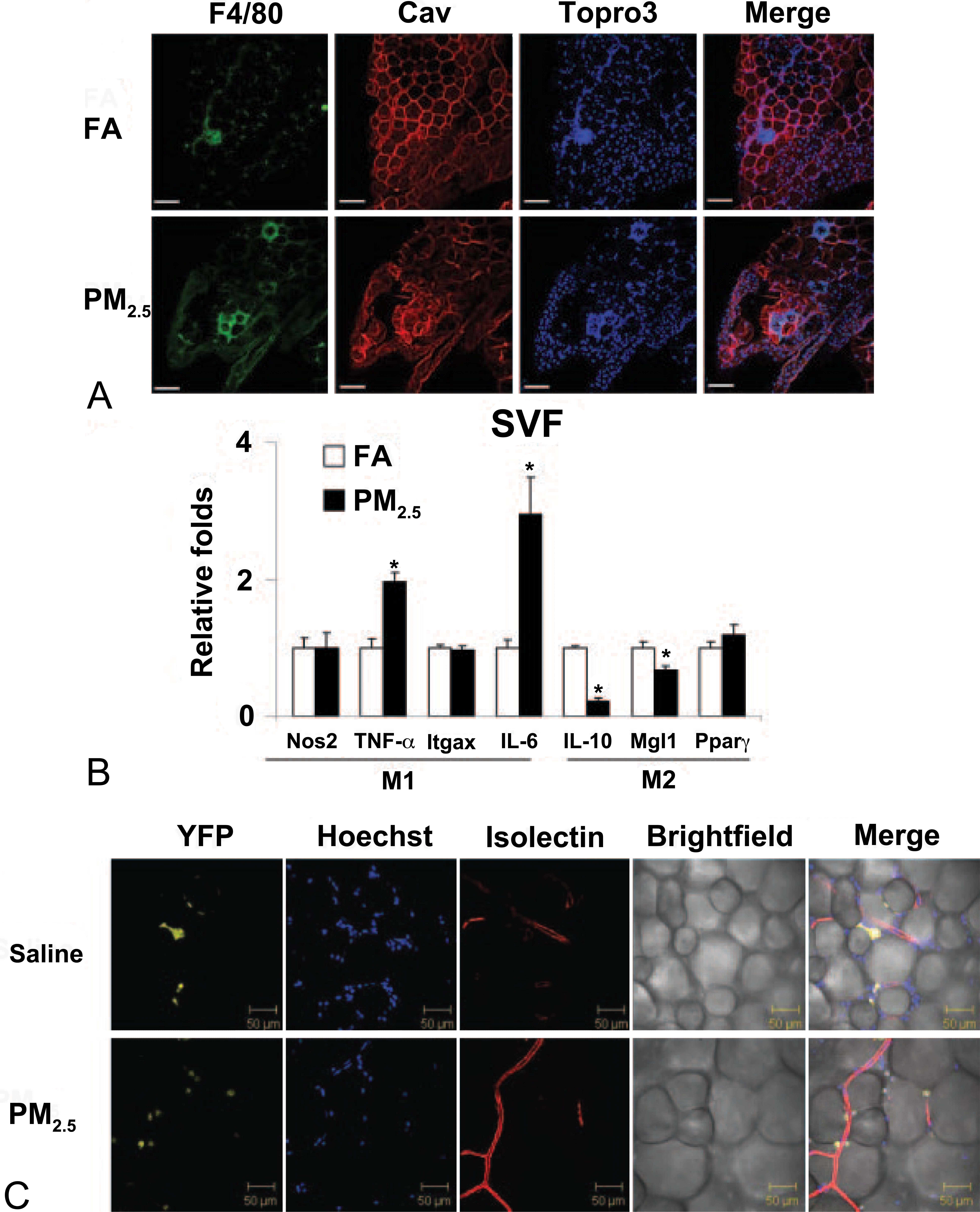
Synergistic effects of diet with high PM2.5 levels. C57BL/6 mice were fed with high-fat diet (HFD; 42% from fat-adjusted calorie diet) and simultaneously exposed to PM2.5 or filtered air. Exposure to concentrated ambient PM2.5 increases macrophage infiltration in adipose tissue and induces a shift in macrophage phenotype in mice fed HFC. A, Immunofluorescence localization of ATMs (F4/80) in epididymal fat pads from C57BL/6 mice exposed to FA or PM2.5. Adipocytes identified by caveolin (Cav) staining and nuclei labeled with TOPRO3. Scale bar = 100 μm. B, Real-time polymerase chain reaction measurement of macrophage M1/M2 gene expression. PM2.5 treatment resulted in significant increases in the M1 phenotypic genes TNF-α and IL-6 in the F4/80+ cells of stromal vascular fraction (SVF). C. Unfixed live adipose tissue from HFC-fed transgenic mice that express yellow fluorescent protein (c-fmsYFP, yellow) was stained with Hoechst 33342 (blue) and isolectin (red) by confocal microscopy. Isolectin is an endothelium-specific marker. PM2.5 treatment resulted in increased YFP cell infiltration into the adipose tissue compared with the saline control. Nos2 indicates nitric oxide synthase-2; Itgax, integrin αX, CD11c; and Pparγ, peroxisome proliferator–activated receptor. *p < .05 vs. FA (Modified with permission from Sun et al., Circulation, 2009.)
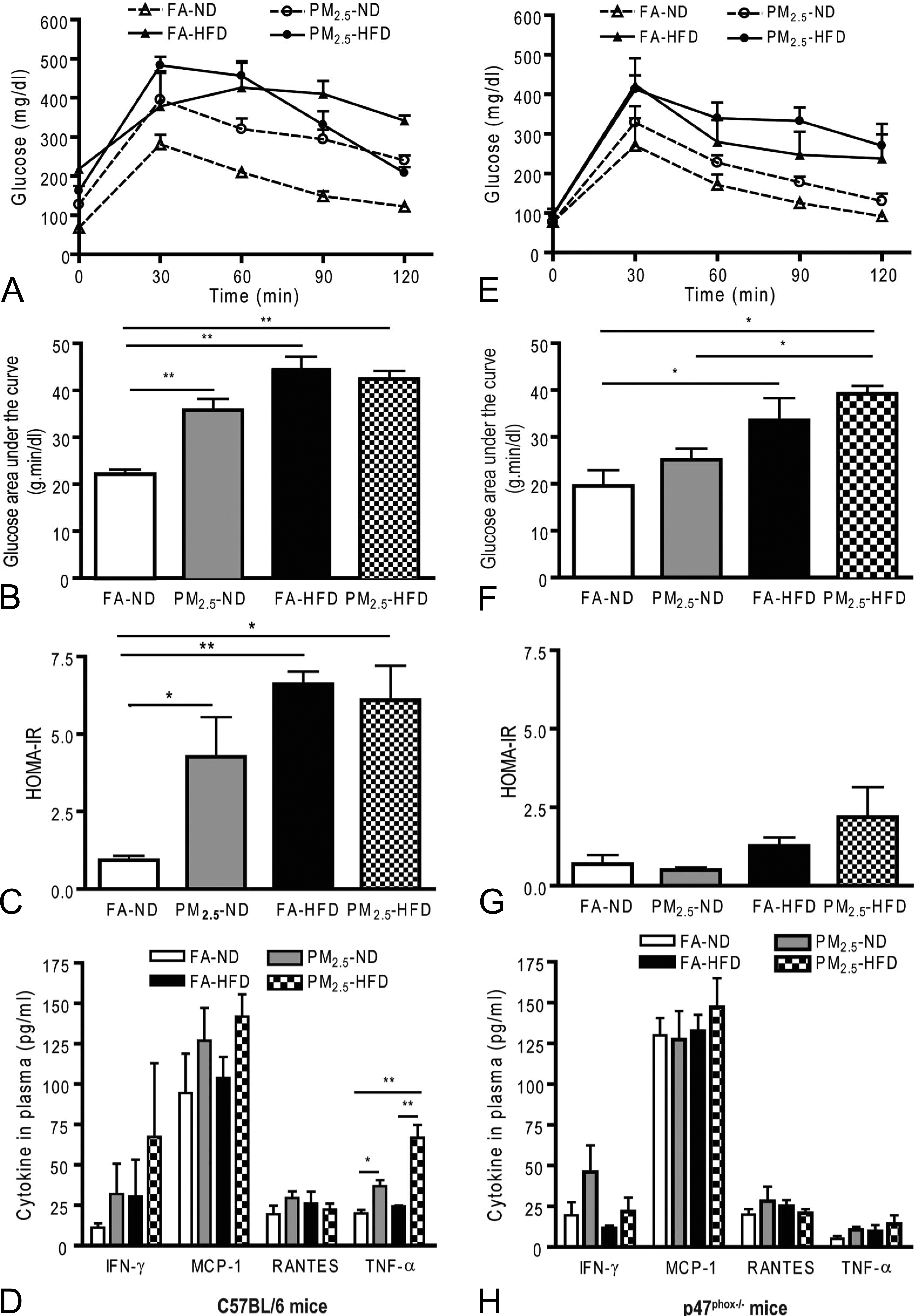
Glucose homeostasis and systemic inflammation in wild-type C57BL/6 mice and p47phox−/− mice by concentrated ambient PM2.5 exposure compared with FA-exposed mice fed a normal diet or a HFD. A and E, Effect of PM2.5 exposure on glucose tolerance by intraperitoneal glucose tolerance test in C57BL/6 mice and in p47phox−/− mice, respectively. B and F, The glucose area under the curve calculated from the
PM2.5-mediated Endoplasmic Reticulum (ER) Stress in Liver and Lung
ER stress, also called unfolded protein response (UPR), is an evolutionarily conserved pathway designed to alleviate protein misfolding in response to diverse physiopathologic stressors (Walter and Ron 2011). In vitro exposure studies have demonstrated that exposure to PM2.5 is capable of inducing ER stress and the UPR and may represent a pathophysiologically relevant mechanism linking PM exposure with hepatic insulin resistance. In in vitro exposure studies, a significant increase in the UPR-associated proteins ATF-4, Hsp70, Hsp90, and binding immunoglobulin protein (BiP) was noted. In response to inhalational exposure to concentrated PM2.5 exposure over 10 weeks, GRP94 (glucose regulatory peptide 94) and BiP were increased in lungs and liver (Figure 4A:1–4), indicating activation of the ATF6 (activating transcription factor 6) pathway in these organs (Laing et al. 2010). ATF6 is one of three key main sensors of ER stress (the others being IRE1a [Inositol Requiring 1a], PERK [double-stranded RNA-activated protein kinase-like ER kinase]). Phosphorylated PERK and eIF2a were also increased in the liver along with an induction of C/EBP homologous transcription factor CHOP/GADD153 (Laing et al. 2010). The latter correlated with apoptosis in the lung and liver. The UPR is known to intersect with a variety of inflammatory and stress signaling systems including the NF-κB and c-Jun N-terminal kinase (JNK) pathways as well as oxidative stress responses, all of which may influence lipid and glucose metabolism. In these studies, a critical role for oxidant stress mediated via NADPH oxidase in activation of the ER stress response was also demonstrated (Laing et al. 2010). The factors involved in the ER stress are summarized as Figure 4B. In subsequent experiments, we have demonstrated that PM2.5 exposure causes a NASH-like phenotype and reduction of hepatic glycogen storage in animals (unpublished data). PM2.5 exposure led to activation of the inflammatory pathway through JNK and downregulation of the IRS1-mediated signaling and peroxisome proliferator–activated receptor (PPARγ2) expression in the liver. These changes were associated with abnormalities in IR and glucose homeostasis.
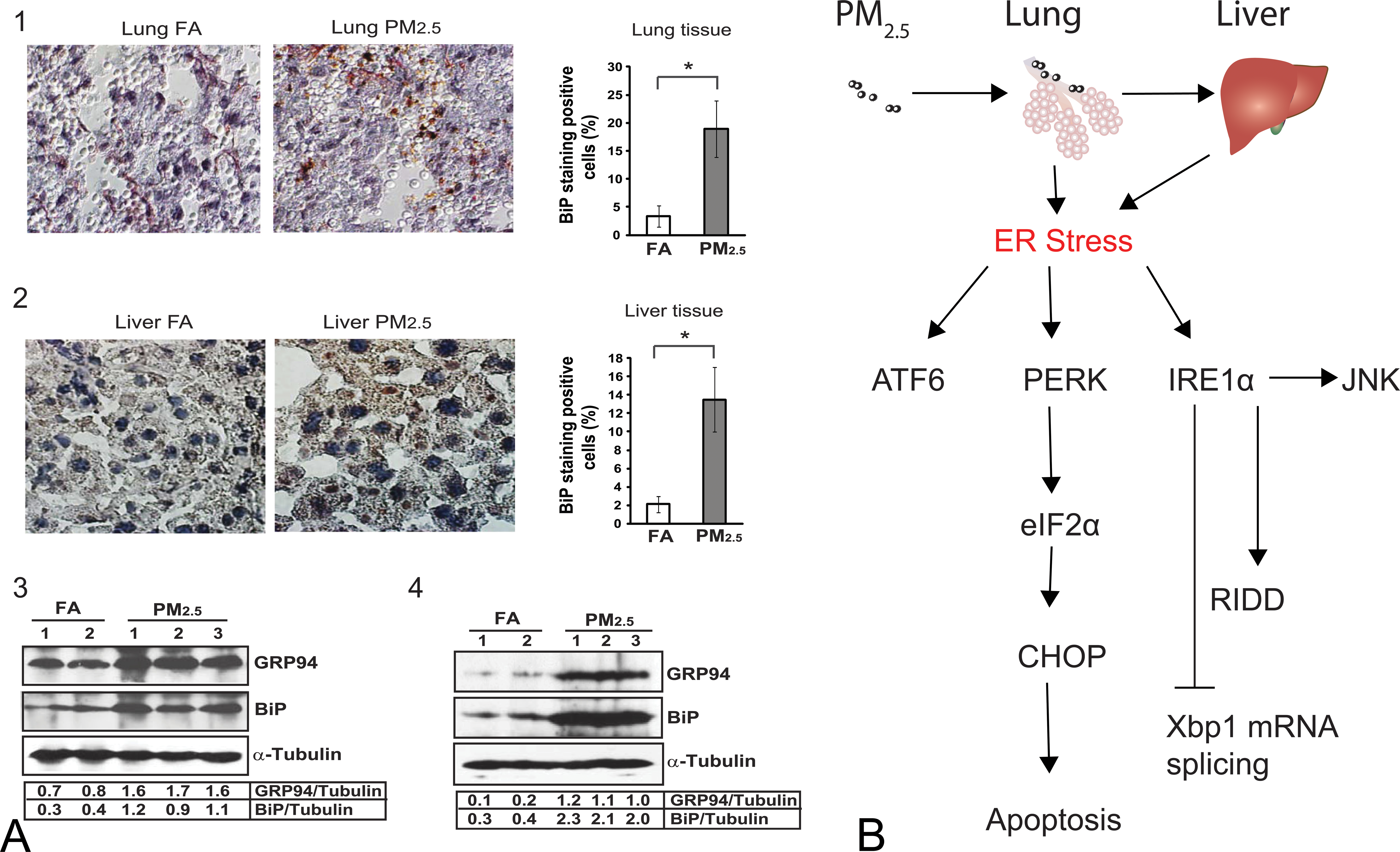
A, PM2.5 exposure induces ER stress in the lung and liver tissues. 1–2: immunohistochemistry staining of lung and liver tissue sections for binding immunoglobulin protein (BiP) expression. The BiP signals were developed with peroxidase substrate reaction (brown signal). The slides were counterstained with hematoxylin. Magnification: ×400. The numbers of positive- and negative-stained cells were counted in 8 random fields per sample. The percentages were calculated by normalizing BiP-staining-positive cells to the total cells. *p < .05 are shown for statistically significant differences. 3–4: Western blot analyses for the expression levels of glucose-regulated protein 94 (GRP94) and BiP proteins in the lung (Brook et al.) and liver (4) tissue of the mice exposed to FA or PM2.5. Denatured lung and liver protein lysates (150 μg per sample for lung and 80 μg per sample for liver) are separated on a 10% Tris-glycine polyacrylamide gel. Levels of α-tubulin protein were determined as internal controls. The values below the gels represent the normalized protein signal intensities. B, a schematic diagram depicting ER stress response pathways induced by PM2.5 in mouse lung and liver tissues. Data were from C57BL/6 mice fed a normal diet and exposed to PM2.5 or filtered air for 10 weeks. (Modified with permission from Laising et al., Am J Physiol Cell Physiol, 2010.)
PM2.5-mediated Mitochondrial Dysfunction and Brown Adipose Tissue (BAT) Dysfunction
Mitochondrial dysfunction has been demonstrated to be a key abnormality in Type 2 DM. Defective fatty acid metabolism through β-oxidation in mitochondria leads to accumulation of intracellular metabolites including fatty-acyl CoA diacylglycerol and ceramide in both skeletal muscle and liver (Lowell and Shulman 2005). We have noted multiple abnormalities in mitochondrial rich BAT with PM2.5 exposure over long durations (10 months) in C57BL/6 mice and over shorter durations (2 months) in ApoE−/− mice results in BAT dysfunction (Xu et al. 2011). Long-term PM2.5 exposure in C57BL/6 resulted in visible decrease in BAT mass with decreased mitochondrial size in BAT depots (Figure 5B). These changes were accompanied by increase in excess oxidative and nitrosative stress in BAT, along with Phase II antioxidant gene induction including NF-E2-related factor 2, NAD(P)H quinone oxidoreductase 1, and glutamate-cysteine ligase modifier subunit. To assess brown adipose dysfunction in more detail, we assessed adipocyte-specific gene profiles in BAT and white adipose tissue by real-time polymerase chain reaction (PCR) analysis. The mRNA levels of the brown adipocyte-specific genes Ucp1, Prdm16, Pgc-1α, and PPARγ2 were significantly decreased in the WAT in response to long-term PM2.5 exposure, although we did not observe any significant difference in C/ebpβ, Cidea, Dio2, or Elovl3 between these 2 groups. In BAT, mRNA levels of Ucp1 and Pgc-1α were significantly reduced in BAT in response to PM2.5 exposure, although there were no significant changes in the other brown adipocyte-specific genes (Figure 5C). Consistent with decrease in Ucp1 gene expression, UCP1 protein was also reduced (Figure 5A). Unpublished observations from our group show that PM2.5 exposure decreased O2 consumption and heat production in a genetic KKay diabetic model. Thus, alterations in BAT may account for the decreased metabolism in response to PM2.5 exposure.
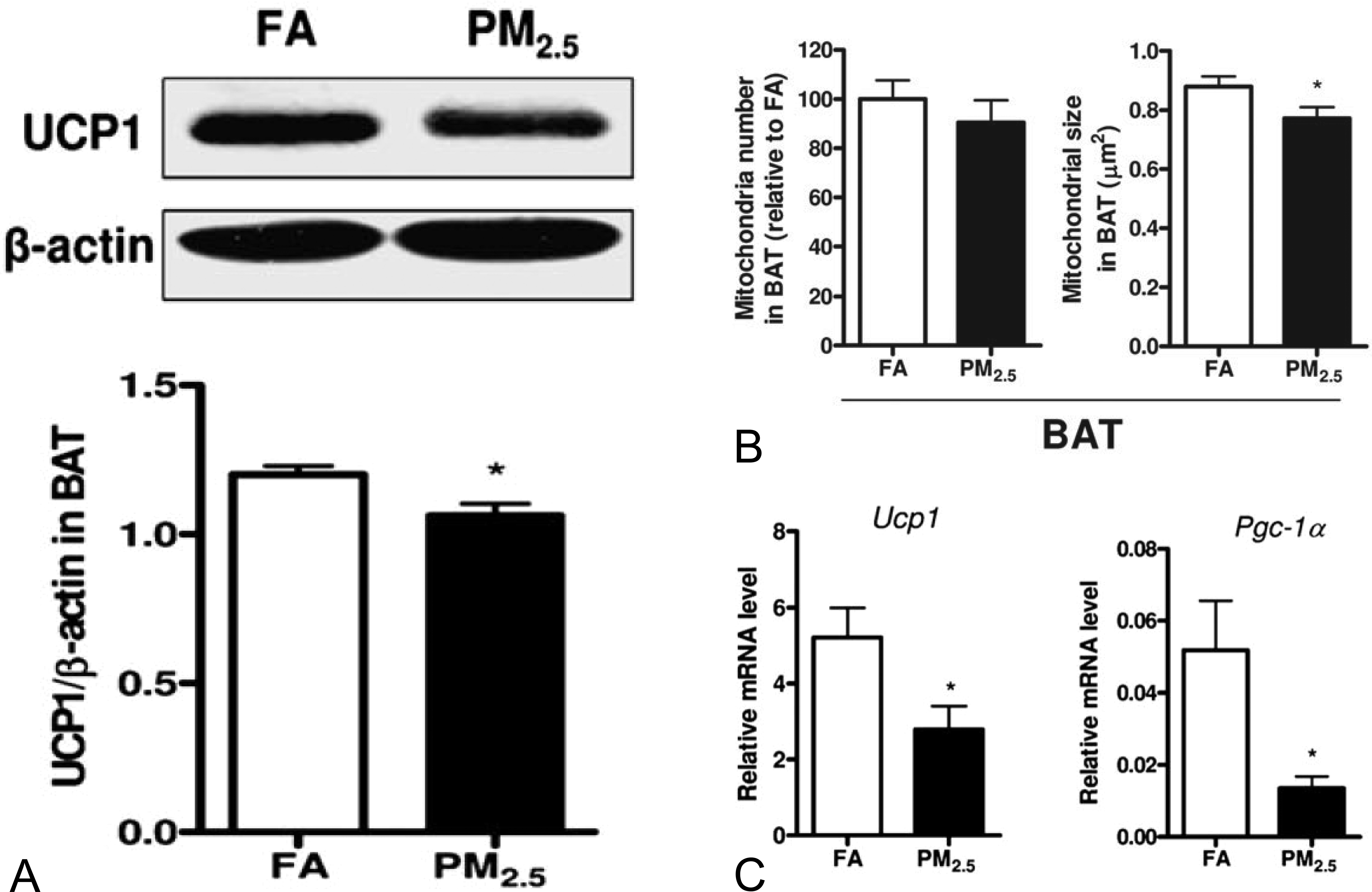
A, Representative bands and statistical analysis of Western blotting for UCP1 expression in BAT in response to PM2.5 exposure. B, Quantification of mitochondrial number and size in BAT in response to PM2.5 exposure. C, Gene expression of Ucp1 and Pgc-1α in BAT in response to PM2.5 exposure. Data were from C57BL/6 mice fed a normal diet and exposed to PM2.5 or filtered air for 10 months. (Modified with permission from Xu X et al, Toxicol Sci, 2011.)
Toll-like Receptors (TLRs) and Nucleotide Oligomerization Domain Receptors (NLRs) as Particulate Matter Sensors
Diet-induced insulin resistance is well known to engage pattern recognition receptors such as TLRs and NLRs. A diversity of ligands such as saturated fatty acids, lipopolysaccharide (LPS) and ceramide have been demonstrated to play a role in both experimental and human models of obesity and insulin resistance (Shi et al. 2006; Vandanmagsar et al. 2011). In contrast to dietary signals, the alveolar macrophage and the bronchial epithelial cells are principal initial cellular sensors of PM and widely express TLRs and NLRs. Biologic components intrinsic to PM such as LPS and peptides and gaseous copollutants such as ozone can directly activate TLRs (Li et al. 2011; Takeda and Akira 2007). Although the level of LPS is lower in PM2.5 versus PM10, there are data linking levels of such components with IR. In a recent prospective study by Sun et al. (2010) in an urban population in Beijing, an important predictor for the development of type 2 DM on multivariate analysis after adjustment of most risk factors including C-reactive protein was LPS-binding protein (LBP). LBP is a better surrogate for LPS in plasma and emerging studies suggest that this may serve as a surrogate for inflammatory disorders resulting from activation of the innate immune system (Lepper et al. 2007; Takeda and Akira 2007). Components such as LPS as part of coarse PM may play a dominant role in urbanized environments in Asia where there may be preferential contamination by sources rich in LPS. The nucleotide oligomerization domain-like receptor Nalp3 has been shown to sense a diversity of particulate components and induce production of interleukin-1β (Dostert et al. 2008).
Endogenous danger-associated molecular patterns (DAMPs) that are released in response to PM may represent additional mechanisms for TLR/NLR activation that may potentiate already overactive pathways in obesity/insulin resistance. In a recent study, a key role for lipotoxicity-associated ceramide accumulation in the pathogenesis of type 2 DM via activation of Nalp3 was demonstrated (Vandanmagsar et al. 2011). A number of DAMPs released in response to PM and/or gaseous components have been demonstrated including oxidized phospholipid components (Figure 6A:1–9) and hyaluronan fragments (Kampfrath et al. 2011; Li et al. 2011). Oxidation products of palmitoyl-arachidonyl phosphocholine (PAPC), an abundant phospholipid in lung lavage fluid, have been implicated in a diverse variety of lung injury signals to activate TLR4 (Imai et al. 2008). Release of oxidized PAPC may facilitate innate immune activation in the lung and function as a mechanism to release chemokines that may then secondarily mediate efflux of inflammatory monocytes from the bone marrow (Kampfrath et al. 2011). We have previously demonstrated that PM2.5 exposure results in egress of CD11b+, Ly6Chi inflammatory monocytes from the bone marrow to circulation and then home to tissue niches such as the perivascular fat via circulation (Figure 6B). These monocytes produce excess amounts of superoxide and may participate in dysregulation of vascular tone. In keeping with these findings, TLR4 deficiency (Tlr4d) or deficiency in the NADPH oxidase subunit Nox2 (Nox2−/−) ameliorated these responses (Figure 6C:1–2) and corrected vasomotor dysfunction. Increased superoxide production required activation of NADPH oxidase as evidenced by increased phosphorylation of the p47 subunit in aortic homogenates in PM2.5 exposed animals, which was prevented by TLR4 deficiency (Figure 6C:3). In parallel in vitro experiments, we demonstrated NADPH oxidase activation by ox-PAPC in cultured bone marrow derived macrophages, an effect which could be prevented by inhibition of interleukin-1 receptor-associated kinase (IRAK), demonstrating that TLR4 mediated IRAK phosphorylation was upstream of NADPH oxidase (Figure 6A-10). Ozone exposure in animal models may mediate degradation of hyaluronan, which can then activate TLR4 via MyD88 pathways (Li et al. 2011). Thus, oxidized phospholipids, hyaluronan fragments, and possibly ceramide as a consequence of air-pollution exposure may represent secondary mediators that may elicit systemic responses. Recent human experiments demonstrate rather rapid inflammatory responses with ozone as evidenced by elevation in interleukin-8 and decrease in plasminogen activator inhibitor-1 at the end of 2 hr of exposure. There was a 104% increase in IL-1β and C-reactive protein levels 24 hr after ozone exposure. The investigators also noted a 51.3% decrease in the high-frequency component of heart rate variability and a 1.2% increase in QT duration compared to pre-exposure levels suggestive of rather rapid autonomic dysfunction (Devlin et al. 2012).
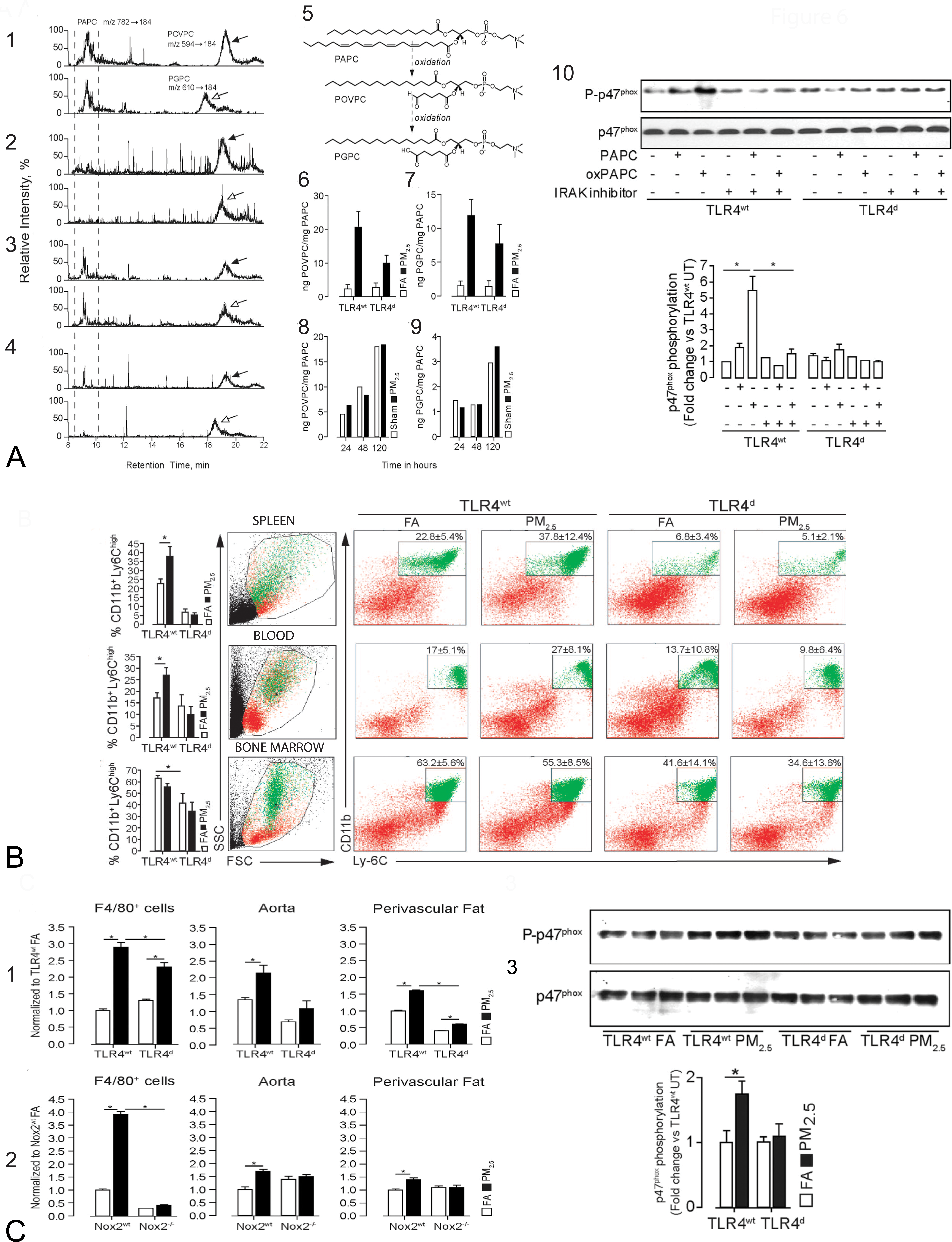
A, Airborne particulate matter causes increased levels of 2 oxidized PAPC derivatives in BAL fluid of PM2.5 exposed mice. Lipid extracts from BAL fluid of TLR4wt and TLR4d mice exposed for 20 weeks to FA or PM2.5 were analyzed by HPLC with positive electrospray ionization mass spectrometry operating in multiple reaction monitoring mode. Parent PAPC and oxidized derivatives (POVPC and PGPC) ion pairs were monitored by their characteristic retention time and daughter ions. Corresponding chromatograms were postprocessed by extraction of POVPC and PGPC ions for quantitative analysis. Representative LC-MS chromatograms are shown for TLR4wt FA (1), TLR4wt PM2.5 (2), TLR4d FA (3), TLR4d PM2.5 (4). 5, Chemical structures of monitored phospholipids. Quantitative analysis of levels of POVPC (6) and PGPC (7) against PAPC with an exaggerated level of oxidation in the PM2.5 exposed mice over 20 weeks. In vitro incubation of PAPC in the presence of PM2.5 or with PBS was performed in a time-dependent manner followed by quantification of levels of POVPC (8) and PGPC (9) by LC/MS-MS.
Central Nervous System Mechanisms in Metabolic Dysfunction: Implications for PM Exposure
Recently, a number of groups have reported inflammation in key regions of the hypothalamus as a mediator of peripheral abnormalities in glucose homeostasis and energy imbalance. Thaler et al. (2012) demonstrated hypothalamic inflammatory signaling as evidenced by upregulation of IL-6 and nuclear factor κB (NF-κB) very early on (within days) prior to substantial weight gain. Furthermore, both reactive gliosis and markers suggestive of neuronal injury were evident in the hypothalamic arcuate nucleus within the first week of high-fat feeding in these experiments. Experiments by Purkayastha et al. (2011) have demonstrated an important role for ER stress in the hypothalamus in the induction of peripheral inflammation and glucose abnormalities. Interruption of ER stress with tauroursodeoxycholic acid (TUDCA) partially reversed obesity-associated metabolic and blood pressure disorders (Purkayastha et al. 2011b). However, acute activation of the proinflammatory protein NF-κB and its upstream activator IκB kinase-β (IKK-β, encoded by Ikbkb) in the mediobasal hypothalamus, an area rich in neurons containing pro-opio melanocortin (POMC), was shown to rapidly elevate blood pressure in mice independent of obesity. Consistent with this, loss-of-function studies of IKK-β selectively in POMC neurons but not Agouti-related peptide neurons counteracted obesity-related hypertension in a manner that was dissociable from changes in body weight (Purkayastha et al. 2011a). These findings may have important implications for potential pathways by which PM2.5 may mediate alterations in peripheral metabolic dysfunction. Particles associated with air pollution have been shown to directly permeate the central nervous system through translocation along the olfactory nerve into the olfactory bulb (Nakane 2012). Alternatively, PM2.5 and/or ozone exposure may directly affect vagal afferents that may play an important role in regulation of pathways that regulate blood pressure or peripheral inflammatory response (Olofsson et al. 2012). Air pollution has been previously shown to cause neuroinflammation, oxidative stress, and pathological alterations such as reactive gliosis (Block and Calderon-Garciduenas 2009). We have previously shown that long-term exposure to PM2.5 over 10 months results in hippocampal pro-inflammatory cytokine expression and impairments in spatial learning memory and behavior (Fonken et al. 2011). Whether PM2.5 exposure results in inflammatory signaling in key hypothalamic centers regulating appetite and neural control of metabolism and inflammation remains to be determined.
Summary
Figure 7 provides a hypothetical framework for mechanism by which inhalational stimuli may cause metabolic dysfunction. Evidence from epidemiologic studies, combined with animal and toxicologic experiments, supports that inflammatory responses to environmental factors is the key mechanism that helps explain the emerging epidemic in cardiometabolic diseases such as diabetes. Both genetic and environmental factors undoubtedly play a role, although the role of the physical and social environment in determining susceptibility may also be critical. Nontraditional factors such as air pollution that are pervasive in the urban environment may provide low level synergism with other dominant factors in accelerating propensity for T2DM. Future studies are warranted to gain greater insight into the molecular mechanisms involved (e.g., intermediary and intracellular signaling pathways), the responsible pollutants (e.g., components, sizes/sources), the role of combined exposures to mixtures (e.g., ozone plus PM), susceptibility factors (e.g., gene–environment interactions, vulnerable populations), and the link of it to the nervous system. Nonetheless, this already important public health issue will likely become of even greater concern in the future, given the current trend toward global urbanization.
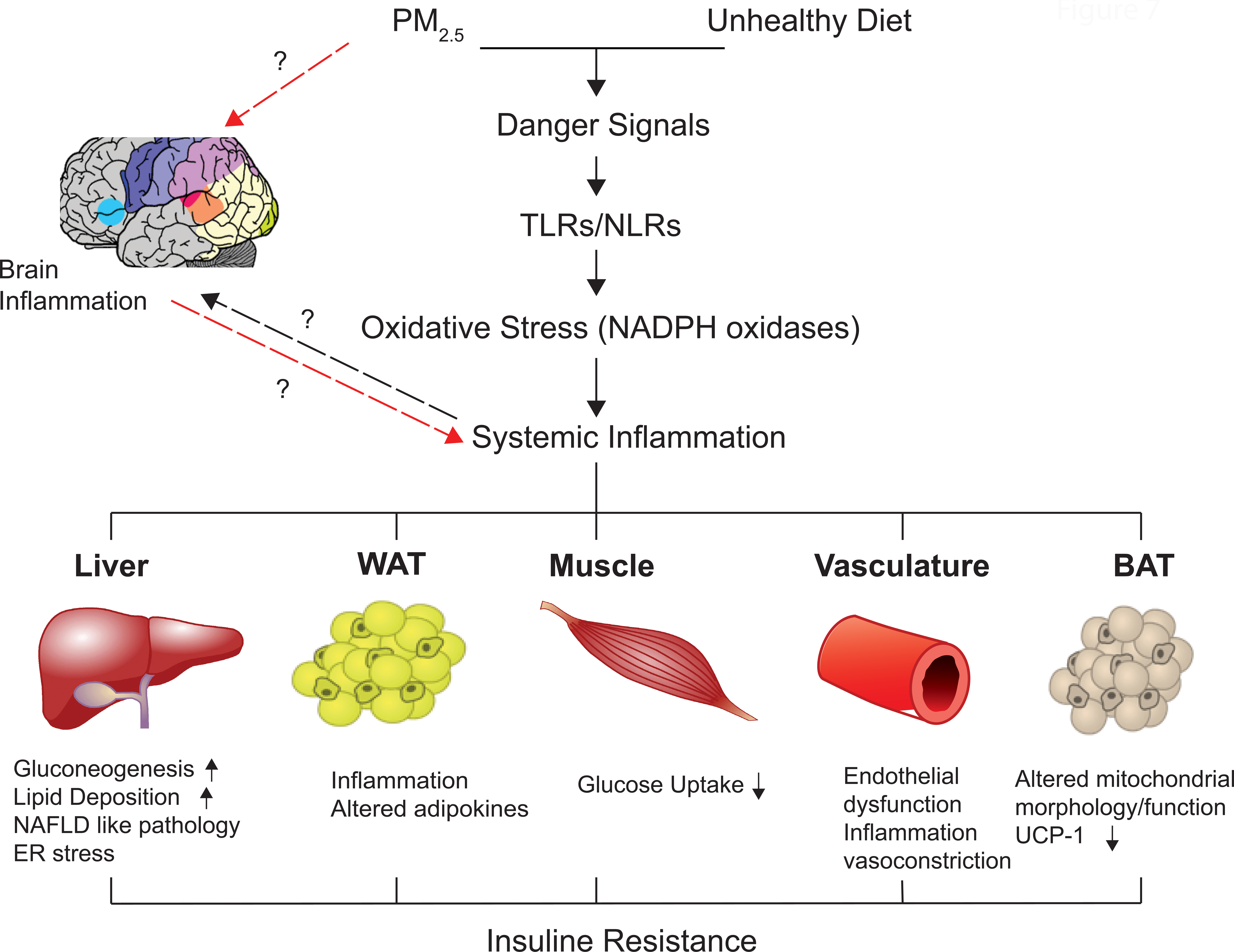
Hypothesized mechanisms of air-pollution-mediated type 2 DM/insulin resistance.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The research was supported in part by NI Grants R01ES017290, R01ES015146 and RO1ES019616 to Dr. Rajagopalan, RO1ES018900 to Dr. Sun, and the U.S. EPA Clean Air Research Center Grant RD83479701 — Great Lakes Air Center for Integrated Environmental Research, to Drs. Rajagopalan, Sun and Harkema.