
Abstract
The Health and Environmental Sciences Institute Cardiac Biomarkers Working Group surveyed the pharmaceutical development community to investigate practices in assessing hemostasis, including detection of hypocoagulable and hypercoagulable states. Scientists involved in discovery, preclinical, and clinical research were queried on laboratory evaluation of endothelium, platelets, coagulation, and fibrinolysis during safety assessment studies. Results indicated that laboratory assessment of hemostasis is inconsistent among institutions and not harmonized between preclinical and clinical studies. Hemostasis testing in preclinical drug safety studies primarily focuses on the risk of bleeding, whereas the clinical complication of thrombosis is seldom assessed. Our results reveal the need for broader utilization of biomarkers to detect altered hemostasis (e.g., endothelial and platelet activation) to improve preclinical safety assessments early in the drug development process. Survey respondents indicated a critical lack of validated markers of hypercoagulability and subclinical thrombosis in animal testing. Additional obstacles included limited blood volume, lack of cross-reacting antibodies for hemostasis testing in laboratory species, restricted availability of specialized hemostasis analyzers, and few centers of expertise in animal hemostasis testing. Establishment of translatable biomarkers of prothrombotic states in multiple species and strategic implementation of testing on an industry-wide basis are needed to better avert untoward drug complications in patient populations.
Keywords
Introduction
Drug safety and efficacy are paramount considerations in the challenging drug development process. These properties are assessed repeatedly in both preclinical (animal) and clinical (human) trials prior to a regulatory agency such as the Food and Drug Administration (FDA) or European Medicines Agency (EMA) granting final permission to market a new medicine. Despite rigorous studies and reviews by pharmaceutical investigators and regulatory agencies to ensure patient safety, some drugs will be recalled from the market voluntarily or by regulatory mandate due to adverse side effects. Cardiovascular complications are one of the primary causes of these marketed drug withdrawals (Schuster, Laggner, and Langer 2005; Redfern et al. 2010). A recent review of cardiovascular safety liabilities further identified that cardiovascular-related attrition of drugs was most prominent during late stage clinical trials and postapproval phases of drug development, and suggested a need for reevaluating the existing preclinical testing strategies of such drugs (Laverty et al. 2011). Many of these cardiovascular liabilities are precipitated by thromboembolic disease (Ageno et al. 2008; Ramot and Nyska 2007). Indeed, the drug withdrawal considered “the biggest safety catastrophe in the United States” (Wadman 2005) is attributed to cardiovascular liability secondary to drug-mediated thromboembolic events. Thus, while drug-induced thromboembolic disease is a very pertinent safety concern, prothrombotic conditions are currently poorly and inconsistently evaluated in drug safety testing, and particularly preclinical assessment. Screening to detect hypocoagulable states predictive of bleeding complications is routinely conducted in preclinical safety studies, but comparable test strategies to detect hypercoagulable states are lacking. As such, thromboembolism is generally detected as a safety concern only after adverse events are identified in clinical patient populations.
There are several recent examples of clinically manifest thromboembolic complications precipitating cardiovascular liability and drug withdrawals. In 2004, 5 years after its approval by the FDA, Vioxx (rofecoxib), a potent Cox-2 inhibitor, was voluntarily recalled from the market due to the increased risk of adverse cardiovascular thromboembolic events including myocardial infarction and stroke (Greener 2005; van Adelsberg et al. 2007; Ramot and Nyska 2007). After scrutiny by the FDA, Celebrex (celecoxib), another Cox-2 inhibitor was required to carry a “black box warning” stating potential risk of adverse cardiovascular events, including myocardial infarction and stroke (Cotter and Wooltorton 2005; Waknine 2010; US FDA 2010). All prescription-strength (non-aspirin) nonsteroidal anti-inflammatory drugs are now required to carry the black box warning label due to a suspected “class effect” of Cox-1 and Cox-2 inhibition (Kuehn and FDA Panel 2005; McKellar, Madhok, and Singh 2007). Safety concerns for serious thrombotic and thromboembolic complications, including cardiovascular adverse events, have been well publicized recently for women taking hormone replacement therapy (HRT) and hormonal contraceptives. This has prompted FDA recommendations to limit HRT duration and to fund an extensive clinical study to compare risk of serious thrombotic and thromboembolic events among hormone contraceptives in order to better understand and limit this risk (Harman et al. 2011; US FDA Drug Safety Communication 2011).
The complications listed above are consistent with drug-mediated abnormalities in one or more components of hemostasis resulting in increased risk of myocardial infarction, vascular thrombosis/thromboembolism, or stroke in susceptible patient populations. Alterations in endothelium, platelets, coagulation, and/or fibrinolysis can all play a critical role in the risk associated with these events (Van Cott and Laposta 2001; Stockham and Scott 2008; Ramot and Nyska 2007).
The regulation of these pathways and laboratory testing of the hemostatic system have been described in detail in excellent reviews elsewhere. Briefly, endothelial cells lining healthy vessels maintain net anticoagulant properties that counteract procoagulant stimuli (Becker et al. 2000; Kwaan and Samama 2010), thereby sustaining blood fluidity. Platelets which are fragments of megakaryocyte cytoplasm and are responsible for primary and secondary hemostasis circulate in an inactive, but rapidly activatable state, and are capable of adhering and aggregating at sites of vascular injury (Boudreaux and Catalfamo 2010). The coagulation cascade of serine proteases and cofactors are kept in check by fluid phase and cell membrane–associated anticoagulant proteins. When the balance between procoagulant and anticoagulant processes is disturbed, the resulting hypocoagulable or hypercoagulable states can manifest as abnormal bleeding or thromboembolic disease that complicate any preexisting conditions (Brummel-Ziedins et al. 2009). The fibrinolytic pathway maintains blood fluidity by initiating the conversion of plasminogen to plasmin which in turn degrades fibrin (Takada, Urano, and Takada 1990). The primary action of the fibrinolytic system is to restore blood flow in occluded vessels (Collen 1980; Erickson et al. 1985).
The preclinical and clinical testing of the major components of hemostasis during the development of new molecules provides biopharmaceutical companies an opportunity to identify abnormalities in hemostasis that may potentially lead to subacute cardiovascular injury and hypocoagulable or hypercoagulable states. However, the current practices for evaluating hemostasis in drug development have seldom been reviewed, nor has a critical analysis been made of these practices in relation to licensed drugs to date. Furthermore, consideration of hemostasis testing in regard to development of protein therapeutics has received little attention. The current testing paradigm for preclinical hemostasis assessment is based on recommendations that are nearly 20 years old (Weingand et al. 1992, 1996). There exists a clear need to develop best practice guidelines and build upon available science to conduct informative and effective safety evaluation.
In an effort to characterize current practices in hemostasis testing and to identify opportunities for improvement, a team of industry, academic, and government scientists participating in the Health and Environmental Sciences Institute’s (HESI) Committee on Cardiac Safety developed a web-based survey in March 2010. HESI is a nonprofit scientific organization that serves as a resource to promote technical collaborations across the scientific community. The primary objective of the survey study was to identify which laboratory tests of hemostasis are routinely conducted or considered in drug development, especially drug safety testing, and the species tested (e.g., mice, rats, dogs, nonhuman primates, ferrets, human beings). The survey was additionally designed to establish for which study stage (regulatory or discovery nonclinical or clinical) the pharmaceutical development community evaluates alterations in the hemostatic system. Information was also collected on the use and perceived utility of “novel” markers of endothelial injury, platelets, coagulation, and fibrinolysis that would provide more value for cardiac safety as translational biomarkers in hemostasis testing during drug development. The results of this survey are discussed in the context of current guidelines and the perceived gaps in testing.
Method
Survey Design and Demographics
The HESI Cardiac Biomarkers Working Group focused this survey on laboratory assessment of hemostasis. Additional evaluation of hemostasis in preclinical drug safety studies includes assessment of clinical signs (live phase data), hematology, gross observations, and microscopic examination of a comprehensive set of tissues in two or more preclinical species.
The web-based survey questionnaire that provided qualitative input to this article was developed by a team of practitioners in the field of hemostasis as part of the HESI Committee on Cardiac Safety’s Biomarker Working Group. The questions addressed how, when, and where alterations in the hemostatic system are assessed during the drug development and drug safety evaluation process. A complete copy of the survey questions is available at http://www.hesiglobal.org/files/public/hesi-hemostasis-survey.pdf. The survey was open for input from March 1, 2010, through May 31, 2010. A link to the online survey was distributed via e-mail to representatives from industry, academe, and government, who were anticipated to have practical experience in the assessment of hemostasis. The survey was hosted by the website SurveyMonkey (www.surveymonkey.com).
Contact names for the distribution list were drawn from the following sources:
Scientists who had expressed interest in participating in the survey or were members of the HESI Cardiac Safety Biomarkers Working Group (these included some of the survey authors). This group included representatives from six different pharmaceutical companies, academe, and the FDA. Scientists who were members of the broader HESI Cardiac Safety Committee (this includes representatives from 18 pharmaceutical companies, 4 academic laboratories, and the FDA and EMA). Scientists from the American Society for Veterinary Clinical Pathology who self-identified as working in the pharmaceutical industry, governmental laboratory, private toxicology service organization, academe, or as a veterinary consultant. Scientists from the American College of Veterinary Pathologists who self-identified as working in the pharmaceutical industry, governmental laboratories, private toxicology service organizations, academe, or as veterinary consultants.
In total, the survey was sent to 275 individuals and 50 responses were returned for an ~18% response rate. Approximately 60 to 80% of survey participants answered at least some questions about endothelium (procoagulant and anticoagulant properties) and approximately 50% answered questions about coagulation and platelets.
About half of the 50 respondents self-identified themselves as representing medium or large size pharmaceutical companies with the remainder affiliated among academe, contract research organizations (CROs), governmental or regulatory agencies, independent consultants/contractors, and scientists from small pharmaceutical companies (Figure 1). Most of the respondents indicated that they participated in preclinical (animal) safety studies conducted according to good laboratory practice (GLP) requirements for submission of data to regulatory agencies. Some respondents (about half) also noted they worked in drug discovery laboratories in lead optimization or non-GLP studies. The response from those participating in human clinical trials was very small and considered insufficient to draw definitive conclusions on clinical testing. Approximately 50% of respondents identified themselves as veterinary clinical pathologists. Toxicologists (26%), veterinary anatomic pathologists (16%), and PhD research scientists (22%) were also represented in the survey. Fewer discovery scientists, laboratory technicians, regulatory scientists, clinical trials physicians, and others participated (Figures 2 and 3).
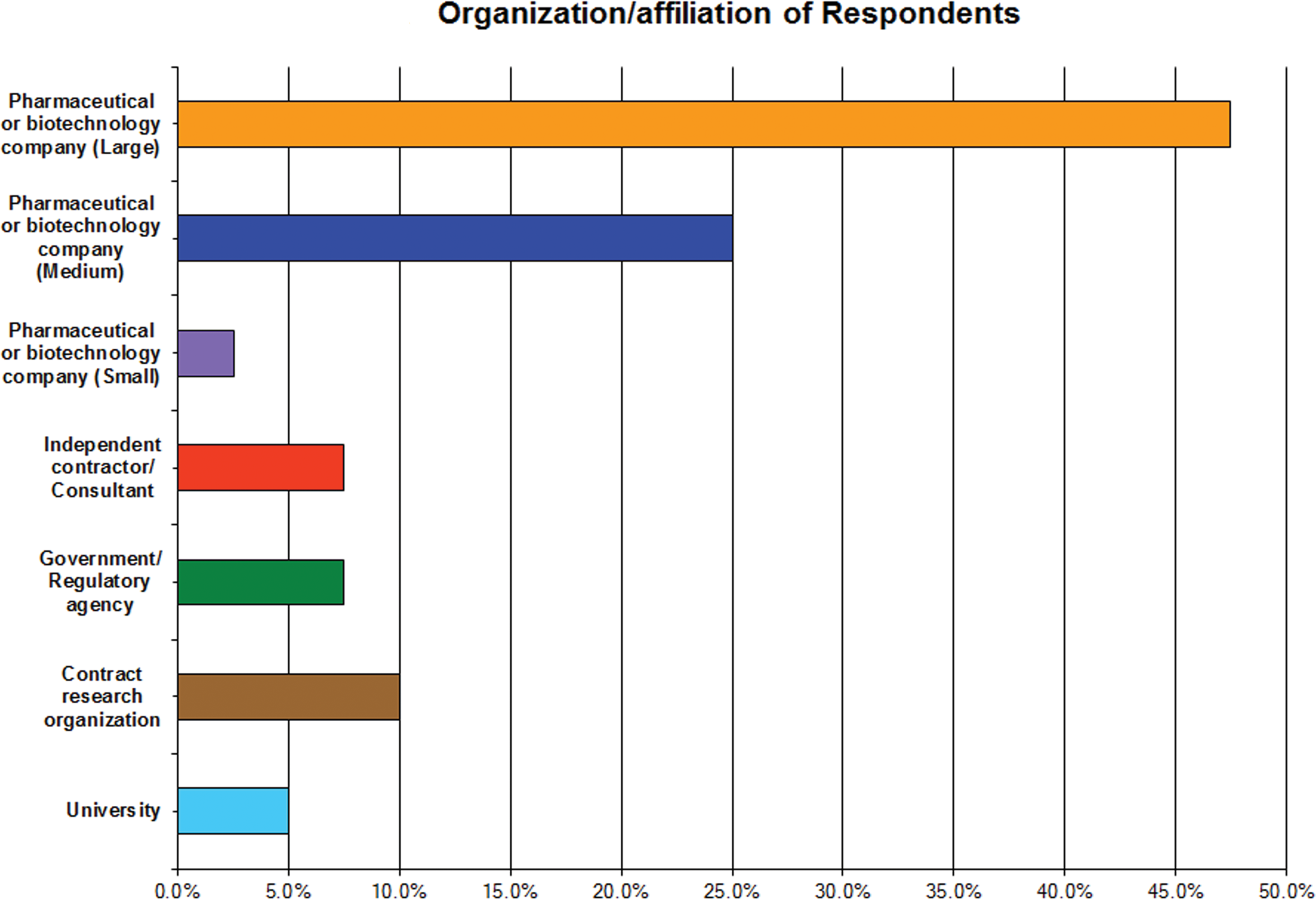
Organizational affiliation of survey respondents. The majority of respondents self-identified themselves as representing medium or large size pharmaceutical companies. Other respondents were affiliated with academe, contract research organizations (CROs), governmental or regulatory agencies, independent consultants/contractors, and scientists from small pharmaceutical companies.
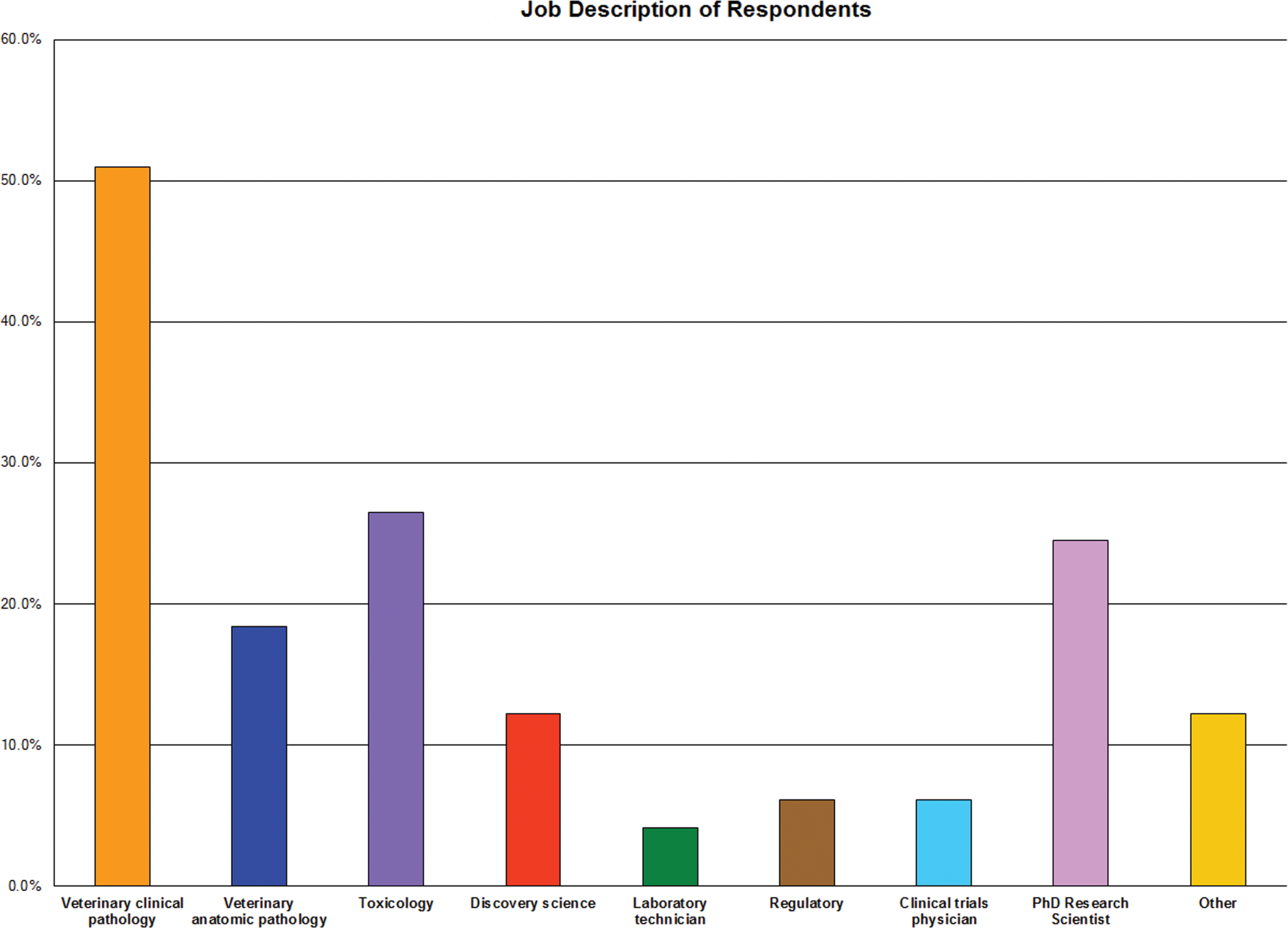
Job description of survey respondents. The majority of respondents identified themselves as veterinary clinical pathologists. Toxicologists, veterinary anatomic pathologists, and PhD research scientists were also represented in the survey.
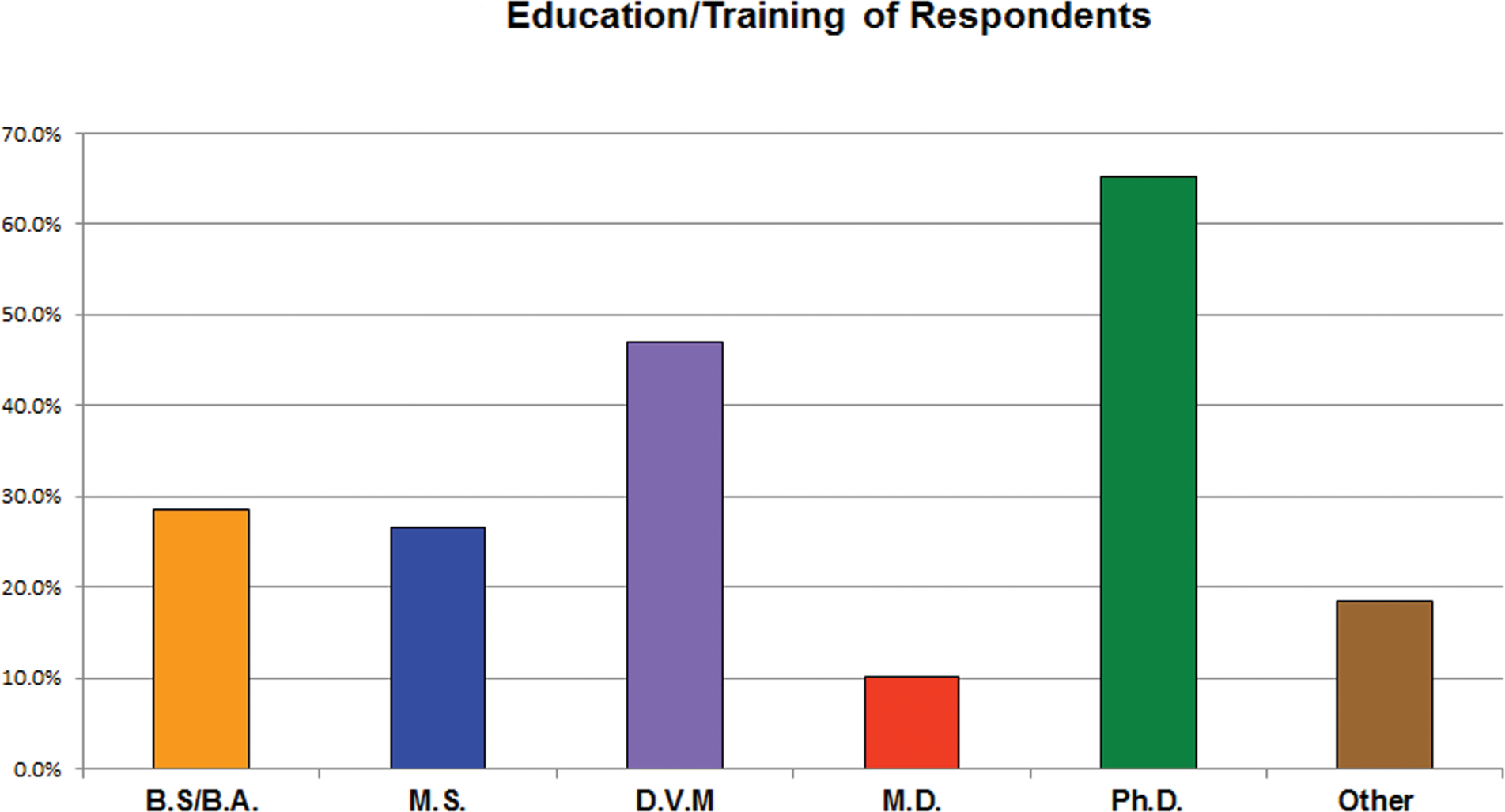
Education and training of survey respondents. The majority of respondents had DVM and/or PhD degrees. Fewer respondents had BS/BA, MS, and/or MD degrees.
While the survey results do not provide definitive data, the number and expertise of the respondents was considered representative of the current approach to hemostasis assessment in discovery and preclinical studies. The total number of respondents engaged in conduct of clinical trials was substantially smaller, and conclusions were accordingly limited and less robust regarding human hemostasis testing in drug development. The survey results are considered sufficient to provide qualitative insights into current trends and perspectives on the laboratory evaluation of hemostasis in preclinical studies.
Results and Discussion
The guidelines for preclinical safety assessment of hemostasis in drug development are derived from a variety of sources and have been reviewed (Hall 1992). Most advisory groups have suggested some measure of clotting competency be performed in animals but have not made a requirement for any specific test. The survey we conducted collected information on the assessment of the four major components of hemostasis: endothelium, platelets, coagulation, and fibrinolysis. The discussion below describes the current guidelines and assessment practices and then compares this with information on the frequency and utility of these practices elucidated by the survey responses.
Published Recommendations for General Hemostasis Testing in Drug Development
Weingand and colleagues (1992), in a joint task force of the American Association for Clinical Chemistry’s Division of Animal Clinical Chemistry and the American Society for Veterinary Clinical Pathology, recommended prothrombin time (PT), activated partial thromboplastin time (APTT), and platelet counts as the minimal data base for laboratory hemostasis testing in animal toxicity and safety studies. The joint task force also described optimal blood collection procedures for use in hemostasis testing in animals. Later, the Joint Scientific Committee for International Harmonization of Clinical Pathology Testing made a similar recommendation of PT, APTT, or alternative, and peripheral blood platelet count for preclinical assessment of hemostasis (Weingand et al. 1996). Notably, tests to assess hypercoagulability or prothrombotic conditions are not mentioned in these recommendations.
Guidance documents describing recommended minimal assessments for hemostasis testing in human clinical trials in drug development are infrequent at best (Theus and Zbinden 1984). These documents, some in the form of national guidelines, provide little detail regarding specific laboratory tests recommended to assess hypocoagulable or hypercoagulable conditions in people that may occur during safety assessment of drugs in development. Health authority guidance documents typically include recommendations for histopathological assessments of tissues. The identification of hemorrhage or thrombosis in an examined tissue section may therefore reflect the only opportunity for assessing hemostatic imbalance. However, routine histopathology is an insensitive means for detection of focal and early thrombotic or hemorrhagic events, and unsuited for characterization of prothrombotic or hypocoagulable states in a live animal. Preclinical studies are typically conducted in healthy animals, where the likelihood of developing widespread histopathological lesions denoting hemostatic imbalance is less than that of clinical patients with underlying disease risk. Thus, histopathology alone is an inadequate means of predicting clinical hemostatic risk and detecting early, nonmorphologic disturbances associated with the coagulation cascade, platelets, fibrinolysis, or endothelium.
To more directly characterize disturbances in the coagulation cascade, a multiparameter panel that evaluates the intrinsic, extrinsic, and common coagulation pathways has been advocated in preclinical GLP studies (Kurata and Horii 2004; Weingard et al. 1992, 1996) and clinical studies (Theus and Zbinden 1984). The APTT, PT, and thrombin time screening tests were designed for identifying deficiencies or inhibitors of coagulation factors that cause ineffective fibrin formation. However, these tests are not capable of characterizing hypercoagulable states. The routine laboratory evaluation of platelets is largely confined to parameters measured by standard hematology analyzers such as the peripheral blood platelet count and indices, whereas platelet reactivity is not assessed routinely. Laboratory assays of fibrinolysis and assessment of endothelial hemostatic properties have not been advocated for routine safety assessment in preclinical testing.
The clinical drug safety community has endorsed the limited common laboratory tests to define adverse events in hemostasis. For example, the United States National Cancer Institute Common Terminology Criteria for Adverse Events (US NCI 2009) relies on the magnitude of change in PT, APTT, fibrinogen concentration, and platelet count to categorize the severity of adverse events in hemostasis, with clinical signs of hemorrhage and thrombosis classifying high-grade events. Notably, the diagnostic performance of even these limited recommended tests to predict hemostatic disturbances (i.e., acting as premonitory biomarkers) has not been systematically evaluated in preclinical species used in safety assessment studies.
Survey Assessment
Survey respondents reported that in the discovery and preclinical stages of drug development, hemostasis evaluations were primarily conducted with healthy animals. A small number of respondents (<20%) utilized animal models of pathologic or induced thrombosis, including rats and nonhuman primates infused with lipopolysaccharide, rabbits with electric current–induced thrombosis, and rats with induced thrombi (method not specified). These studies were considered to potentially improve modeling and enhance detection of complications that might be encountered in a clinical population.
Among preclinical test species, dogs, nonhuman primates, and rats were considered acceptable models for hemostasis testing. Mice were not considered acceptable models for hemostasis testing due to blood volume constraints and spurious influences of difficult phlebotomy. Hemostasis testing was not likely to be performed in the ferret, an animal used by some institutions to investigate cardiac safety. Approximately 80% of respondents cited a lack of confidence in hemostasis test results in one or more preclinical species used at their institutions.
Survey participants were asked to identify specific indices or analytes considered successful translational biomarkers of hemostatic pathways for use in human clinical trials. Cited most frequently by respondents to this question were the PT (76%), APTT (76%), platelet count (59%), and fibrinogen concentration (41%). Platelet aggregation (29%),
For coagulation testing with GLP studies including those at CROs, PT, APTT, and fibrinogen concentration were most often conducted. Nonroutine tests of coagulation were reported to be infrequently conducted by the CROs. Tests for endothelial procoagulant or anticoagulant properties and for fibrinolysis were reported to be performed only rarely by CROs. The survey respondents reported that the assessment of platelet end points at CROs was routinely conducted as part of clinical studies more than nonclinical studies. Positive responses pertaining to CRO testing for platelet function were similar to, or lower than those for in-house routine GLP or discovery nonclinical testing. Respondents reported a relatively greater frequency/scope of platelet end points evaluation in CRO-conducted clinical studies relative to in-house clinical studies, a difference believed to reflect greater overall reliance on CROs for clinical study conduct in industry.
Tests of Endothelium
Tests of endothelial hemostatic properties were divided into procoagulant (prothrombotic) properties or anticoagulant (antithrombotic) properties (Table 1). The vast majority of respondents reported that only limited testing was conducted for endothelium-associated procoagulant markers (e.g., TF [tissue factor], vWF [von Willebrand Factor], PAI-1 [plasminogen activator inhibitor–1], Factor V). Testing was performed to investigate mechanisms for specific abnormalities, most often in rat studies and less frequently in the dog, primate, mouse, human, and ferret. Respondents indicated that testing for the endothelial anticoagulant properties (negative endothelial surface charge, PGI2, NO, TM, TPA, TFPI, and CD39) was performed even less frequently. Respondents were unlikely to pursue these assays even to investigate mechanisms for specific findings with compounds. Reasons for not using these end points included the lack of species-specific assays for endothelial markers of procoagulant and anticoagulant properties and the scarcity of laboratory facilities that offer these tests.
Tests of endothelial hemostatic properties.

Tests of Platelets
Tests of platelet hemostatic properties were divided into quantitative and qualitative or functional tests (Table 2). Most respondents indicated that one or more quantitative evaluations of platelets (peripheral blood platelet count, mean platelet volume [MPV], platelet distribution width [PDW], reticulated platelet count, or mean platelet component concentration [MPC]) occurred routinely in hemostasis assessment in preclinical and clinical drug development studies. Survey respondents generally indicated satisfaction that analysis of these quantitative end points of platelets was adequately covered in drug safety studies.
Tests of platelet hemostatic properties.

Platelet function tests were not assessed routinely in pre-clinical or clinical drug safety testing. Rather, respondents indicated these assays were limited to investigative/discovery studies or on an as-needed basis in regulatory studies. In general, platelet function testing was listed more frequently for clinical trials than preclinical studies. Platelet aggregation testing was considered an investigative parameter in studies with rats, dogs, and nonhuman primates (but not mice or ferrets).
The use of platelet reticulocyte counts in nonhuman primates, platelet surface–associated immunoglobulin (PSAIg) and antiplatelet antibody in dogs, and the platelet contribution to clot strength (thrombelastography [TEG]) and blood thromboxane B2 in human beings were also cited as assays considered for investigative studies. The lack of broad-based assessment of platelet function, and insufficient laboratory evaluation of platelet–endothelium interactions were considered important gaps in preclinical testing.
Coagulation
Survey participants were asked about a variety of tests of coagulation (Table 3). Tests routinely assessed in GLP preclinical (animal) studies to support human dosing of drugs in development include APTT, PT, and fibrinogen concentration. These tests were only sometimes assessed in human clinical trials.
Tests of coagulation.

In preclinical, discovery and GLP studies, the current practice is to conduct routine coagulation screening tests in healthy animals. Results of these routine tests in preclinical studies were considered by most respondents (75%) to be translatable to the clinic. However, a lack of cross-species correlative data for alterations in coagulation tests to predict the magnitude and significance of a change for clinical populations was repeatedly noted. Mice and ferrets were the only preclinical species for which coagulation test results were considered poorly translatable to the clinical population. The major gaps in coagulation testing for preclinical and clinical studies were the lack of sensitive and specific tests for detecting hypercoagulability in all species. End point times (PT, APTT) or concentrations (fibrinogen) are also species-specific, and hence the translation to the clinic or across species cannot be made by laboratory-defined cutoff values.
Fibrinolysis
Survey participants were asked about a variety of tests of fibrinolysis, antiphospholipid syndrome, and anticoagulant proteins (Table 4). The relative number of responses to questions relating to fibrinolysis was generally low (16–42% of all survey participants). The survey responses indicated that tests for fibrinolysis and coagulation inhibitors were not performed routinely in regulatory or discovery studies for preclinical or clinical phases of drug development. Only investigators involved with human clinical trials indicated they conduct fibrinolysis testing. The following markers were considered appropriate as investigative tests to characterize a specific compound or to investigate mechanisms of action on an individual drug basis: fibrin and fibrinogen degradation products (FDPs),
Tests of fibrinolysis, antiphospholipid syndrome, and anticoagulant proteins.

Gaps in Hemostasis Testing
The most commonly noted gap in laboratory testing of hemostasis in both preclinical and clinical studies by the survey respondents was the lack of appropriate tests to detect hypercoagulability/prothrombotic states. For preclinical safety assessment, the requirement for relatively large sample volumes to conduct hemostasis testing and the lack of cross-species reactivity of immunologic tests for proteins involved in hemostasis were also considered major limitations. Individual comments cited concern about the lack of general coagulation testing in exploratory rat and mouse studies, and the uncertain clinical translation of the magnitude for drug-induced changes in preclinical species in PT or APTT. Approximately 22% of respondents cited no gaps or concerns with current hemostasis testing in preclinical species at their institutions.
Similar to the concerns expressed for preclinical studies, respondents to questions on clinical studies cited the lack of accurate tests for hypercoagulability/prothrombotic states (30%) as the most common gap. Also mentioned was the lack of requirement for hemostasis testing in routine clinical studies (i.e., often hemostasis testing is done only in response to a related clinical finding; 15%), heterogeneity of performance of clinical coagulation tests (specifically in the APTT) and absence of routine systematic evaluation of platelet function in drug development. Approximately 38% of respondents to the direct question noted no gaps or concerns with current hemostasis testing in clinical studies.
Conclusion
This study reports on results of a survey of the pharmaceutical development community pertaining to hemostasis testing for nonclinical and clinical drug safety evaluation. Notably, the tests of hemostatic assessment discussed in this article are only one part of a broader spectrum of cardiovascular safety evaluation. However, considering the contribution of hemostatic imbalance to cardiovascular disease, and that cardiovascular disease is the predominant cause of both human mortality and drug withdrawals worldwide, attention to this system in drug safety testing of human-use drugs seems particularly prudent.
Results of this survey indicate the evaluation of hemostasis in drug safety testing is largely nonstandardized among institutions and nonharmonized between nonclinical and clinical studies. Results also confirm that hemostasis testing in routine drug safety studies is generally focused on the risk of bleeding rather than the clinical risk of thrombosis. The survey responses indicated that with the exception of coagulation screening tests, functional assessments of hemostasis (e.g., endothelial and platelet interactions, fibrinolytic pathways) are not undertaken to inform safety risk. Respondents identified gaps in hemostasis testing that include the lack of availability and implementation of tests for the evaluation of prothrombotic states. The lack of information on the predictive value of preclinical test results for clinical events, and on the most appropriate preclinical models for evaluating specific hemostasis risks were also indicated. None of the survey respondents or discussants mentioned concerns with current assessments for bleeding potential.
Ultimately, the best strategy for hemostasis testing in drug safety is contingent upon recognition of the gaps in the current testing paradigm, selection, and qualification of translatable biomarkers of hemostasis in multiple species with tactical and practical implementation on an industry-wide basis. Post hoc evaluation of the outcome of implementing additional or different tests will determine the effectiveness of this strategy for improving cardiovascular risk assessment. Based on results of this survey, the authors believe there is a gap in the preclinical laboratory testing for the detection of prothrombotic and hypercoagulable states that warrants investigation of new or different tests by the HESI Cardiac Biomarkers Working group. The goal of new tests will be particularly focused on improving risk assessment of prothrombotic conditions that might predispose to myocardial infarctions, thrombosis/thromboembolism, and/or stroke. The gaps and opportunities listed above will be the focus of the HESI Cardiac Biomarkers Working Group in the near future.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.