
Abstract
Exposure to air pollution has been associated with acute myocardial ischemia, impaired myocardrial function, and ST-segment depression. Particulate matter (PM)–associated metals, especially vanadium and nickel, have been implicated in observed cardiovascular impairments. We aimed to assess the effect of single intratracheal pulmonary exposure to vanadium-rich respirable oil combustion PM (HP-10) on the intrinsic myocardial ischemic tolerance and mitochondrial integrity in rats. The authors subjected isolated heart tissue slices derived from saline or PM-exposed rats to low glucose low oxygen induced ischemia followed by oxygenated condition with glucose supplementation. Mitochondrial structural integrity was determined by TEM (transmission electron microscopy) and functionality by the 3-(4, 5 dimethylthiazol-2yl)-2, 5 diphenyltetrazolium bromide (MTT) assay. Rats exposed to PM exhibited no apparent inhibition of mitochondrial dehydrogenase activity in oxygenated conditions at 24 or 48 hr post–PM exposure. However, in conditions of simulated ischemia/reoxygenation, these heart slices showed a delayed but consistent and significant decrease in dehydrogenase activity compared to controls at 48 hr after exposure to PM. Electron microscopy revealed significant myocardial mitochondrial injury upon exposure to PM characterized by mitochondrial swelling and fusion. The authors conclude that exposure to soluble vanadium-rich PM induces mitochondrial functional impairment and structural abnormality, which compromises mitochondrial respiration and results in decreased tolerance to ischemia/reoxygenation in rats.
Introduction
It has been estimated that long-term exposure to air pollution increases the risk of cardiovascular morbidity by 24% and of cardiovascular mortality by 76% for every 10 µg/m3 increase in PM2.5 (Miller et al. 2007). The estimated annual premature mortality due to air pollution by the World Health Organization reaches 0.8 million (Mills et al. 2009; http://www.who.int/whr/2002/en/whr02_en.pdf, p. 69). Poloniecki et al. (1997) have shown a direct association between events of acute myocardial infarction (MI) and previous day air pollution in London, England. Zanobetti and Schwartz (2007) showed that PM inhalation is not only associated with triggering MI, but also with increased incidence of death, recurrent MI, and development of heart failure following MI. Hu and Rao (2009) have shown the association between chronic ischemic heart disease (IHD) and particulate air pollution in the eastern United States. Even short exposures to air pollution have been associated with marked increases in cardiovascular morbidity and deaths from myocardial ischemia, arrhythmia, and heart failure, especially among patients with underlying coronary artery disease (Hassing et al. 2009; Hu and Rao 2009; Pope et al. 2002, 2006). A recent statement of the American Heart Association affirmed that the evidence concerning the association between PM2.5 exposure and cardiovascular morbidity and mortality is consistent with a causal relationship (Brook et al. 2010).
Air pollution components, such as transition metals, have been associated with risk factors of ischemia, atherosclerosis, and hypertension, and have been implicated in direct effects on vascular reactivity, coronary structure, and thrombogenesis in humans and experimental animals (Floyd et al. 2009; Harrabi et al. 2006; Sun, Yue, Kirk, et al. 2008; Sun, Yue, Ying, et al. 2008; Suwa et al. 2002). Although few animal studies have shown cardiac conductance abnormalities after PM exposure, the evidence of molecular alterations leading to contractile dysfunction is less well understood (Gottipolu et al. 2009; Kodavanti et al. 2011). More specifically, PM-associated vanadium and nickel have been linked to adverse cardiovascular outcomes (Sun, Yue, Kirk, et al. 2008; Sun, Yue, Ying, et al. 2008; Suwa et al. 2002). We have shown that leachable PM-associated transition metals translocate rapidly to the circulation and are found in the heart following pulmonary exposure (Wallenborn et al. 2007). In case of zinc, cardiac gene expression changes following pulmonary exposure reflect direct effect of zinc on the myocardium (Gilmour et al. 2006a, 2006b). However, the experimental evidence remains inconclusive about biochemical changes that are associated with cardiac physiological impairment following exposure to PM containing vanadium and nickel.
The outcome of cardiac ischemic events depends not only on the size, intensity, and duration of the ischemia but also on the intrinsic defense mechanisms of the myocardium (Ferdinandy et al. 2007; Golomb et al. 2009; Peart and Headrick 2008; Ravingerova 2007; Schwartz and Sack 2008). Cardiac ischemic tolerance is dynamic; constantly modulated by metabolic, neuroendocrine, paracrine, and autocrine stimuli; and regulated via various signal transduction pathways (Ferdinandy et al. 2007; Ravingerova 2007) affecting mainly myocardial mitochondria (Schwartz and Sack 2008). Therefore, ischemic injury can be viewed, at least partially, as a failure of the mitochondrial defense mechanisms (Golomb et al. 2009). Exogenous agents may injure the heart by decreasing its ischemic tolerance (Golomb et al. 2007, 2009).
The aim of the present study was to assess the effect of pulmonary exposure to PM, which contained leachable vanadium on cardiac ischemic tolerance and mitochondrial integrity. For this purpose, we administered a respirable oil combustion fly ash PM sample rich in water-soluble vanadium (HP-10); collected from a power plant in Boston, MA), by intratracheal instillation into rats (Kodavanti et al. 1998). We used an ex vivo technique to simulate ischemia in isolated cardiac tissue sections (Ad et al. 2005; Golomb et al. 2009, 2007). The viability of the myocardium was then examined in vitro, in oxygenated conditions and in simulated ischemia/reoxygenation (I/R). Mitochondrial structural alterations were examined by electron microscopy.
Materials and Methods
Animals
Male Sprague-Dawley rats (Harlan Laboratories, Israel) were grown in SPF pathogen-free conditions. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (www.nap.edu/catalog/5140.html). The joint ethics committee (IACUC) of the Hebrew University and Hadassah Medical Center approved the study protocol for animal welfare.
PM Intratracheal Instillation and Ex Vivo Experiments
The PM sample HP-10 is a preparation of vanadium-rich oil combustion particles collected from a power plant in Boston, MA (Kodavanti et al. 1998). This oil fly ash particle preparation contained 35.6 mg/mg water-leachable vanadium and 0.7 mg/mg water-leachable nickel (kindly provided by Dr. David Christiani and Dr. Russ Hauser, Harvard School of Public Health, Boston, MA). To understand the temporality of PM effects on the heart, initially ischemic tolerance was studied only with high concentration of PM (2.5 mg/kg body weight) but at two time points (24 and 48 hours postinstillation). In a second set of experiments, PM concentration-related effects were examined at 48 h postinstillation using 0.5 or 2.5 mg/kg dose-levels.
Healthy male rats (300 ± 20 g) were anesthetized with ketamine-xylazine, intubated, and ventilated. The ventilation was disconnected, and 0.3 of saline, or suspended HP-10 in saline, was instilled into the trachea through intubation tube, as described by Kodavanti et al. (1998). The rats were connected to the ventilator for an additional 5 min to ensure efficiency of the intratracheal instillation. Twenty-four or 48 hours later, heparin sodium, 500 units, was injected (i.p.) into the rats to avoid blood coagulation. Thirty minutes later, rats were anesthetized with sodium pentobarbital (i.p. 30 mg/rat). Hearts were immediately removed and placed in heparinized ice-cold saline solution.
Ex Vivo Experiments and MTT Assay
The use of rat ventricular sections in an ex vivo experimental system simulating ischemia and reoxygenation (I/R) was previously described in our earlier studies involving isolated human right atrial sections (Schneider et al. 2003) and rat left ventricular (LV) sections (Golomb et al. 2007). Briefly, 2 millimeter cubes from the midsection of the LV were exposed to 30 min of oxygenated equilibration in 25 ml of glucose supplemented phosphate buffered saline (G-PBS) bubbled with oxygen at 37oC (O2-G-PBS), then washed in PBS and transferred for 90 min to conditions of simulated ischemia: PBS without glucose bubbled with nitrogen (N2-PBS, at 37oC), and then to 90 min of reoxygenation with glucose (O2-G-PBS, 37oC), in the presence of MTT obtained from Sigma (St. Louis, MO, USA). Oxygenated control cubes from each LV were incubated in O2-G-PBS throughout the entire experiment. Glucose-phosphate buffered saline (G-PBS) was prepared freshly and used as our incubation medium for the in vitro experiments. The buffer consisted of (mM): NaCl 136.9, KCl 2.68, Na2HPO4 8.10, KH2PO4 1.53, MgCl2 6H2O 0.5, CaCl2 2H2O 0.9, glucose 5.55 (pH 7.45). The buffer of the simulated ischemia consisted of the same ingredients without glucose.
At the end of each experimental protocol, the reduction of MTT to blue formazan by mitochondrial dehydrogenases in the tissue was assessed (Bes et al. 2004). The sections were exposed to MTT for the entire period of reoxygenation—5 sections were incubated in 20 ml O2-G-PBS containing MTT (0.5 mg/ml) at 37oC for 90 min. The sections were then transferred to a small test tube containing 3 ml saline, which was shaken for 1 min to remove excess dye. Then, the sections were wiped on a gauze cloth and transferred to 15 ml plastic test tubes and frozen overnight. Extraction of the Formazan dye into 10 ml of dimethyl sulfoxide (DMSO) was done with vigorous shaking for 1 hour at 37oC. The absorbance of the colored supernatant was measured on a spectrophotometer at 500 nm. To determine the dry weight, the sections were dried in a 90oC oven for 24 h and weighed.
The MTT results obtained in oxygenation or following I/R protocols are expressed as optical density (OD) per mg dry weight of myocardial tissue. The ratio between the optical densities following I/R to that of its oxygenated controls, normalized to dry weight of the myocardial tissue, reflects the myocardial tolerance to ischemia/reoxygenation, and is referred to as the ischemic tolerance index (ITI) (Golomb et al. 2007).
Histopathology—Tissue Collection and Processing
Forty-eight hours following the intratracheal instillation the rats were anesthetized with pentobarbital, the heart, aorta, and lungs were removed and fixed in 4% neutrally buffered formaldehyde for histopathological examination. Particular care was taken to inflate the lungs with formalin in order to get the normal inspiration volume. Tissues were trimmed, embedded in paraffin, and routinely processed for light microscopy. Sections were stained with hematoxylin and eosin (H&E). A board-certified toxicological pathologist (A.N.), blinded to the treatment, performed the histopathological evaluation.
A semiquantitative grading scheme was used to evaluate the extent of the lesions, generally using the criteria presented by Shackelford et al. (2002), using 5 grades, as follows: no lesion (grade 0), minimal (grade 1), mild (grade 2), moderate (grade 3), and marked (grade 4).
Examination of Heart Samples by Transmission Electron Microscopy (TEM)
Ultrastructural changes of the heart specimens were assessed using electron microscopy by an experienced pathologist (CC) blinded to the study group. Samples of heart tissue, taken 48 hours after HP-10 instillation, were trimmed into approximately 1-mm3 strips and placed into 0.1 M Sorensen’s sodium phosphate buffer (pH 7.2). The samples were postfixed in 1% osmium tetroxide, rinsed in distilled water followed by dehydration in a series of graded alcohols then acetone. Finally, the samples were embedded in Epon-812 epoxy resin (Electron Microscopy Sciences, Hatfield, PA) and allowed to polymerize overnight at 60oC (six blocks each/animal). Following polymerization, the blocks were trimmed, a 0.5 µ thick section (semithin) was cut, stained with 1% Toluidene blue, examined by light microscopy, and then two blocks were chosen for thin sectioning. Thin sectioning consisted of an ultrathin, 80–90 nm, “gold” section placed on a 150 µ mesh thin-bar copper grid then stained with uranyl acetate and lead citrate and examined on a JEOL 1011 transmission electron microscope at 80kV. The ultrathin sections were most representative of all the semithin samples for that particular animal. Twenty digital photomicrographs were taken at 10,000 ×/animal (two grids were made/10 micrographs/grid: each grid representing a separate piece of tissue and each micrograph represented a different region on that particular grid). Each animal’s photomicrographs were evaluated separately, then compared by group. Lung and cardiac tissues were also processed for histology and examined at light microscopy level.
Statistical Analysis
Data are expressed as mean ± standard deviation. Statistical significance of differences between groups was determined using ANOVA and Tukey’s post hoc test. A p-value less than .05 was considered significant.
Results
We aimed to examine whether pulmonary exposure to PM, which contained leachable vanadium, would cause acute myocardial alterations and mitochondrial impairment. To test this, we administered a respirable oil combustion fly ash PM sample rich in water-soluble vanadium (HP-10; collected from a power plant in Boston, MA), by intratracheal instillation into rats (Kodavanti et al. 1998). We used an ex vivo technique to induce ischemia in isolated cardiac tissue sections (Ad et al. 2005; Golomb et al. 2009, 2007). The viability of the myocardium was then examined ex vivo, in oxygenated conditions and in simulated ischemia/reoxygenation (I/R). At the concentrations used (0.5 or 2.5 mg/kg), PM exposure led to pulmonary pathology (Figure 1A and B) consistent with what has been observed in other studies (Kodavanti et al. 1997). Multifocal minimal to mild (i.e., grade 1 to 2) intra-alveolar accumulation of dark pigment-laden macrophages was noted in the lungs of the animals exposed to HP-10 2.5mg/kg. Occasionally, the adjacent alveolar lining epithelium was hyperplastic. In addition, scarce polymorphonuclear (PMNL) cells were sporadically associated with the histiocytes (Figure 1A and B).
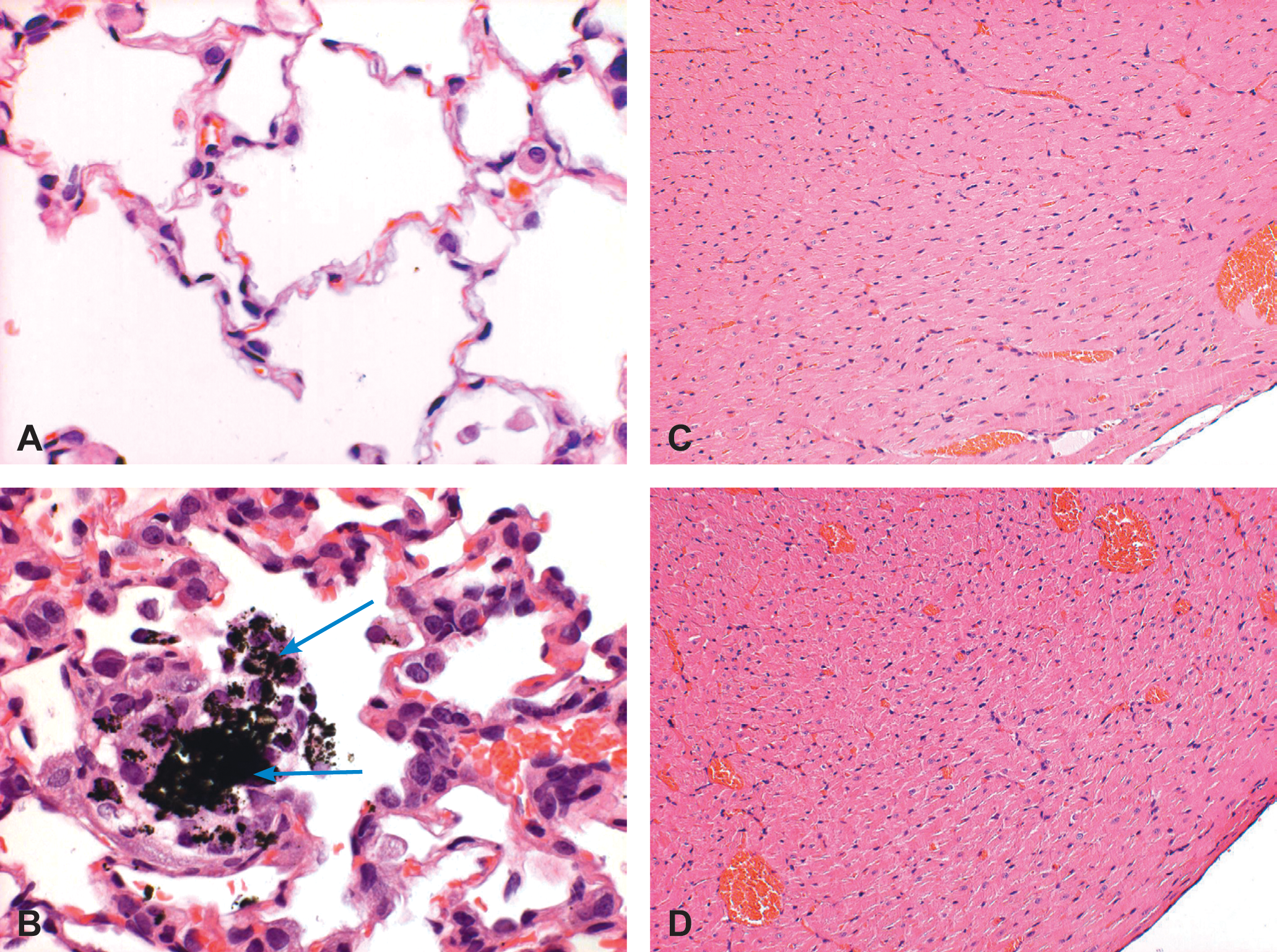
Histology of the lungs and heart after instillation of HP-10. (A) Histological section of the lungs from a control animal (H&E). (B) Histological section of the lungs from a rat, 48 hours after exposure to HP10 2.5mg/kg intratracheally. Note intra-alveolar accumulation of dark pigment-laden macrophages (arrows). Occasionally, the adjacent alveolar lining epithelium was hyperplastic (H&E). (C) Histological section of the heart left ventricular myocardium from a control animal. Note, no abnormalities are seen (H&E). (D) Histological section of the heart left ventricular myocardium, from a rat exposed to HP10 2.5mg/kg intratracheally. Note no abnormalities are seen (H&E).
Myocardial Redox Activity Is Preserved in Oxygenated Conditions Following PM Instillation
Left ventricular sections of the heart exposed to the concentrations used (0.5 or 2.5 mg/kg) showed no difference in their morphology (Figure 1C and D). To test the effects of PM on myocardial redox activity, rats underwent intratracheal instillation with two doses of HP-10 (0.5 or 2.5 mg/kg) or control saline, and left ventricular (LV) sections were isolated. We found that 24 and 48 hours after exposure to 0.5 mg/kg or 2.5 mg/kg of HP-10 there was no apparent effect on MTT reduction in LV sections without simulated ischemia and with adequate supply of oxygen and glucose (O2-G-PBS) (Figure 2). No abnormalities were detected in the heart after PM instillation under experimental conditions containing adequate supplies of oxygen and glucose.
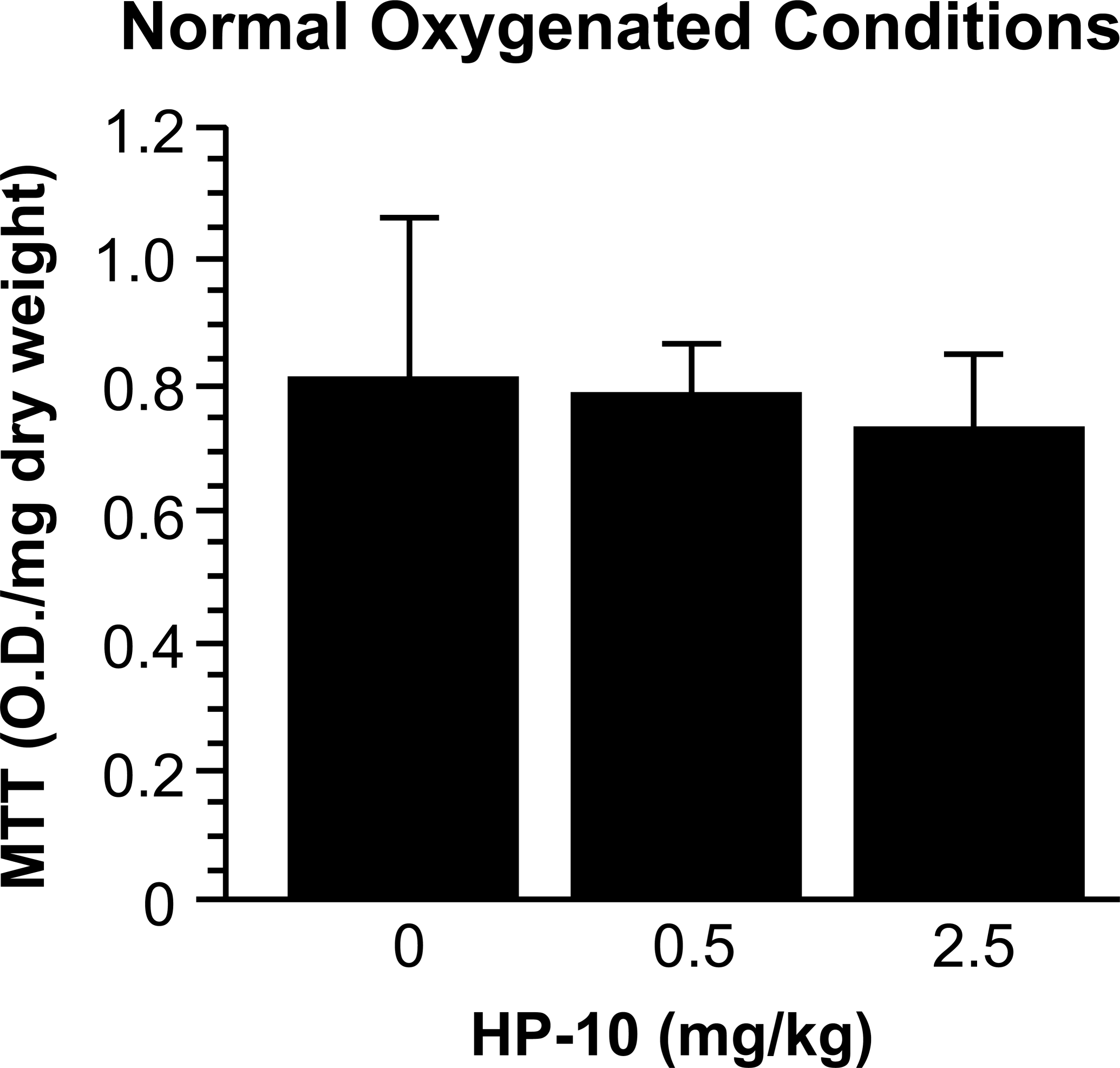
Normal myocardial mitochondrial function in oxygenated conditions after PM instillation. During pre-ischemia (oxygenated control), MTT activity in LV myocardial sections from rats instilled with HP-10, 0.5 or 2.5 mg/kg (n = 8 and n = 6, respectively; 48 hours after exposure to HP-10) did not differ from the activity measured in untreated rats (saline control = 0, n = 7). O.D. = Optical density at 500 nm.
Dose- and Time-Related Cardiotoxicity of HP-10 during Ischemia/Reoxygenation (I/R)
Since myocardial recovery following ischemia has been shown to be a sensitive indicator of myocardial impairment, we employed ischemia protocol by exposing cardiac tissue slices to low oxygen and low glucose during incubation followed by reoxygenation and glucose supplementation. Twenty-four hours after exposure to 2.5 mg/kg of HP-10, there was no apparent change in myocardial ischemic tolerance (Figure 3) as determined by reduction of MTT as a result of mitochondrial succinate dehydrogenase activity. In contrast, MTT activity in LV sections incubated for 48 hours in conditions simulating I/R from rats exposed to 2.5 mg/kg of HP-10 was significantly reduced compared to control (a 34.7% decrease), p < .05 (Figure 4A). Thus, myocardial ischemia tolerance index (ITI) decreased significantly (p < .05) 48 hours after exposure to 2.5 mg/kg of HP-10 (Figure 4B). There was no measurable effect of exposure to lower dose (0.5 mg/kg) of HP-10 on myocardial ischemic tolerance after either 24 or 48 hours (Figure 4).
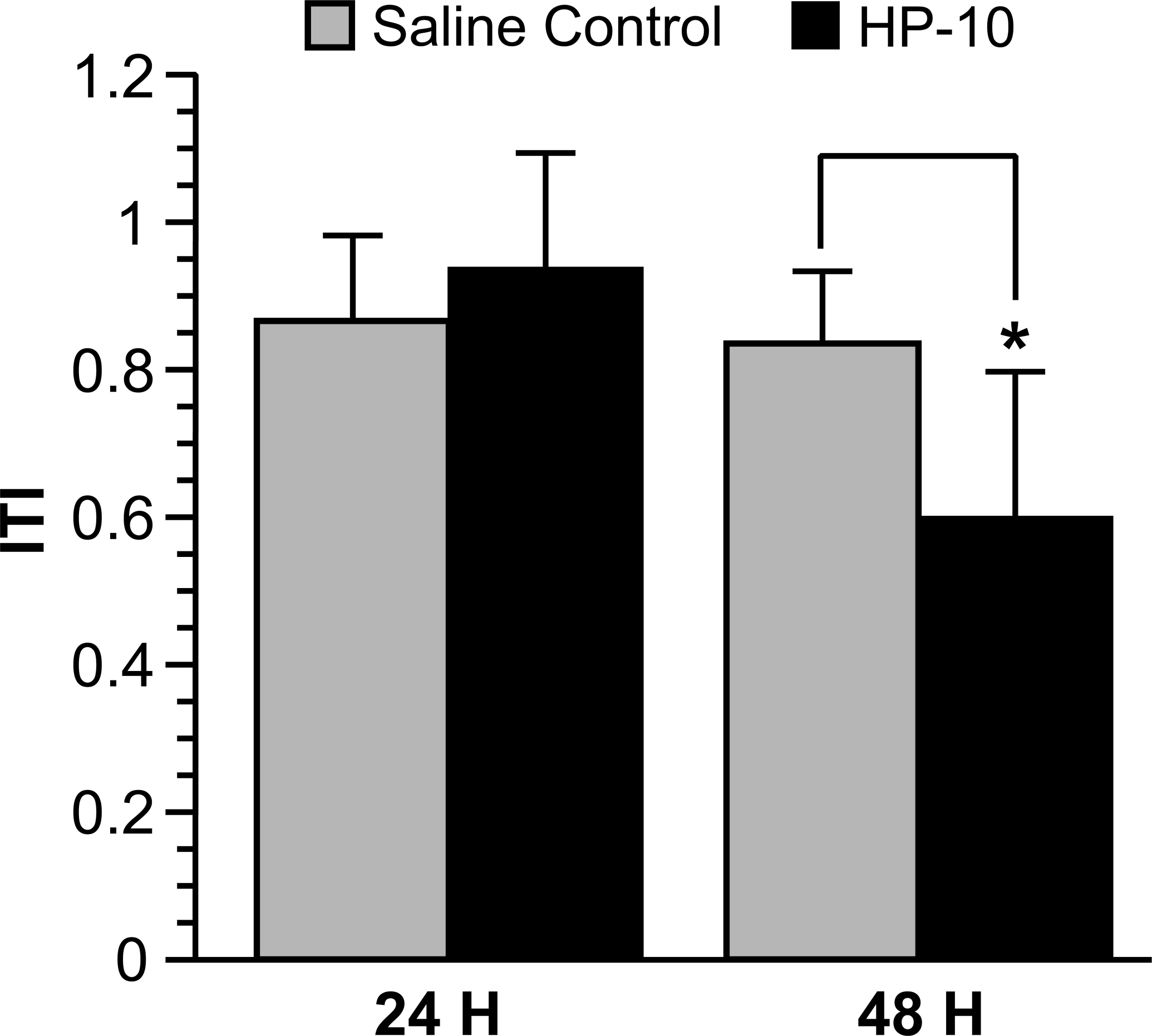
Time-dependent cardiotoxicity by HP-10 instillation. Ischemic tolerance index was calculated from MTT results of myocardial LV sections obtained 24 hours or 48 hours after rats were instilled with saline control or HP-10 (2.5 mg/kg). Ischemia was simulated by depleting glucose in the media and incubating cardiac slices under depleted oxygen conditions. The figure shows a significant decrease in ITI in the HP-10 instilled hearts (*p < .05, n = 4) following 48 hours of the instillation of the HP-particulate matter.
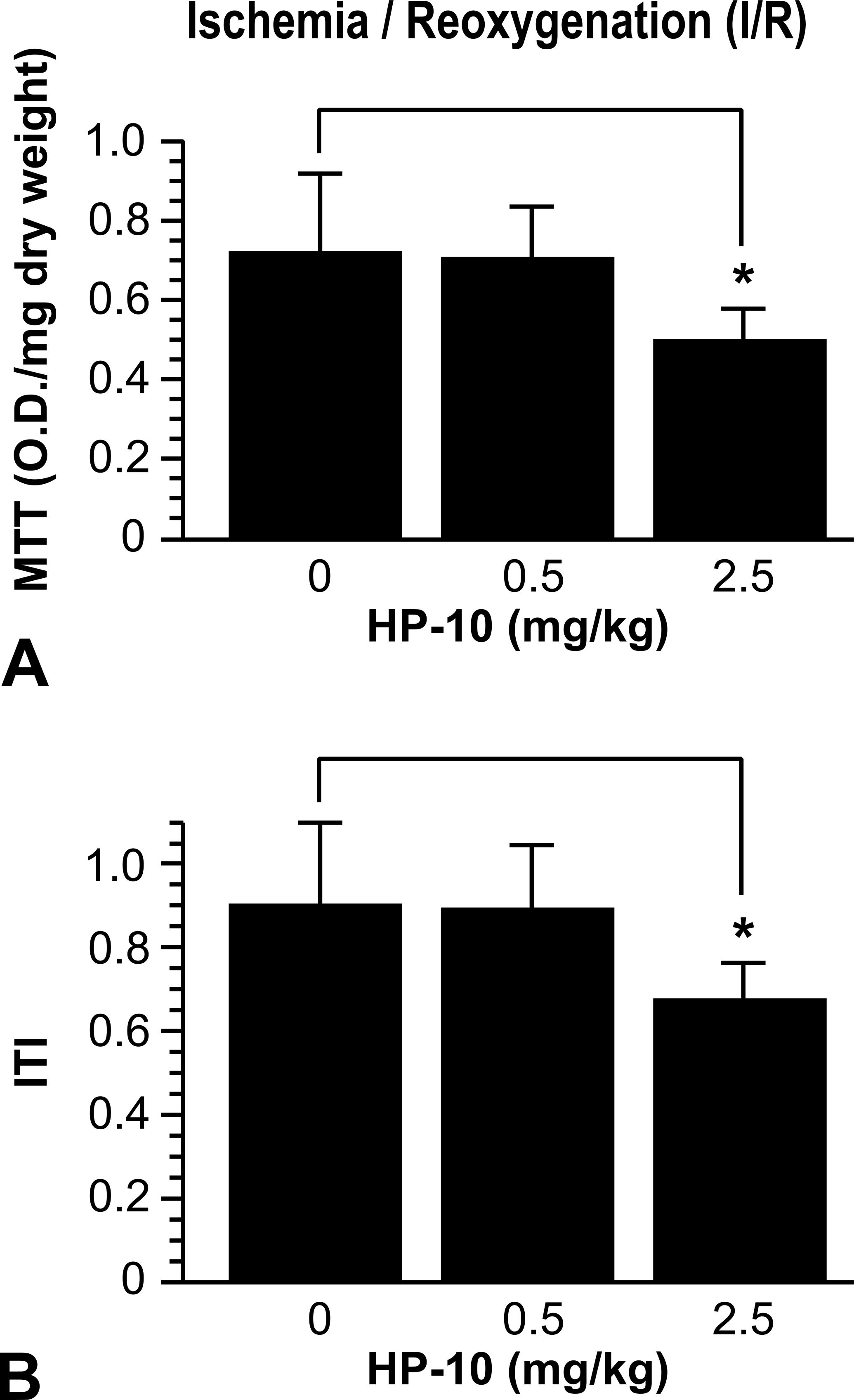
HP-10 instillation causes decreased myocardial redox activity during simulated ischemia/reoxygenation (I/R). (A) LV sections were obtained and MTT assay was performed as described in Figure 1, 48 hours after instillation of HP-10, in simulated ischemia/reoxygenation (I/R). Instilled 2.5, but not 0.5, mg/kg HP-10 significantly decreased MTT reduction (following I/R) versus the untreated saline controls (*p < .05). (B) Ischemic tolerance index (ITI) was measured in simulated I/R in myocardial sections treated as described in Figure 1. The figure indicates a significant decrease in ITI in the instilled 2.5 mg/kg HP-10 hearts (*p < .05).
Cardiac Mitochondrial Injury Induced by Instillation of HP-10: Ultrastructural Analysis
To determine whether the PM-induced impairment in mitochondrial response to I/R conditions reflects a structural injury, we examined myocardial ultrastructural morphology. As shown above, examination of LV section using light microscopy revealed no apparent changes in myocardial morphology after exposure to low or high dose of PM (Figure 1C and D). In contrast, by electron microscopy, we found that PM instillation caused a marked increase in mitochondrial swelling, fission, or partitioning (septation) of the inner mitochondrial membrane (IM), as well as an increase in fusion of multiple mitochondria producing giant mitochondria and megamitochondria (Figures 5 and 6). We observed increased fragmentation, disorganization/disorientation, clumping, and/or loss of mitochondrial cristae following PM instillation. Normal cristae formation, such as longitudinal/linear pattern, was not evident after PM instillation. Instead, in rats exposed to PM, we observed vesicular clumps, fragmented mitochondrial cristae as well as concentric whorls, swirls, and concave loops (Figures 5 and 6). In addition, PM exposed myocardial samples revealed increased quantity and magnitude of mitochondrial fission and fusion compared to controls (Figures 5 and 6). The contents of the sarcoplasm and the myofibrils were also different in PM-treated animals compared to controls (Figures 5 and 6). Subjectively, there was an increase in randomly scattered lipid droplets, lysosomes, and glycogen particles within the sarcoplasm of cardiac cells in the treated animals compared to the cardiac cells of the control animals. Overall, the difference in mitochondrial swelling, the characteristics of the cristae, as well as the appearance of the sarcoplasm were markedly different compared to controls, 48 hours after exposure to 2.5 mg/kg of PM, indicating a significant mitochondrial injury.
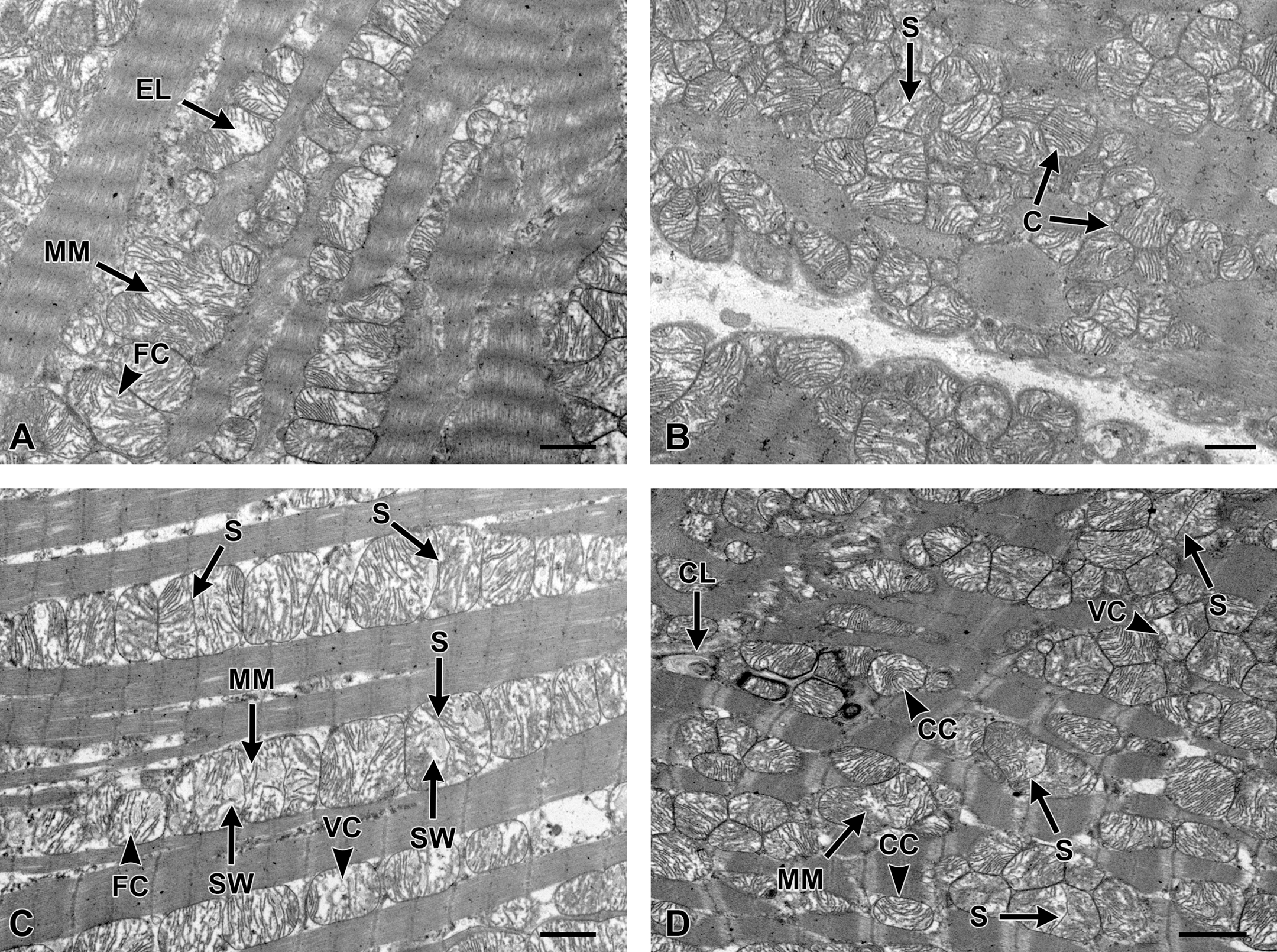
Photomicrographs illustrating ultrastructural features between control (A, B) and particle treated heart samples (C, D) obtained 48 hours after HP-10 instillation. Moderate mitochondrial swelling in control heart samples (A) and (B) was observed. The majority of mitochondria were less rounded and more polygonal shaped than in control samples. Unlike in control samples, where the mitochondrial cristae are closely packed, uniform, and linear, in 2.5 mg/kg PM, these mitochondrial cristae [C] were expanded, fragmented [FC], or completely missing, leaving an electron lucent [EL] matrix. In image (B), there is one mitochondrion showing prominent fission or partitioning [septation, S] of the inner membrane. In 2.5 mg/kg PM treated heart samples, images (C) and (D), there was an increase in fusion [megamitochondria, MM] and mitochondrial fission [S]. Typical mitochondnria damage had associated damage to the cristae such as fragmentation [FC], occasional concaving of cristae [CC], cristae forming swirls [SW], concave loops [CL], and/or clumps of vesiculated debris [VC]. Bar indicates 2 microns.
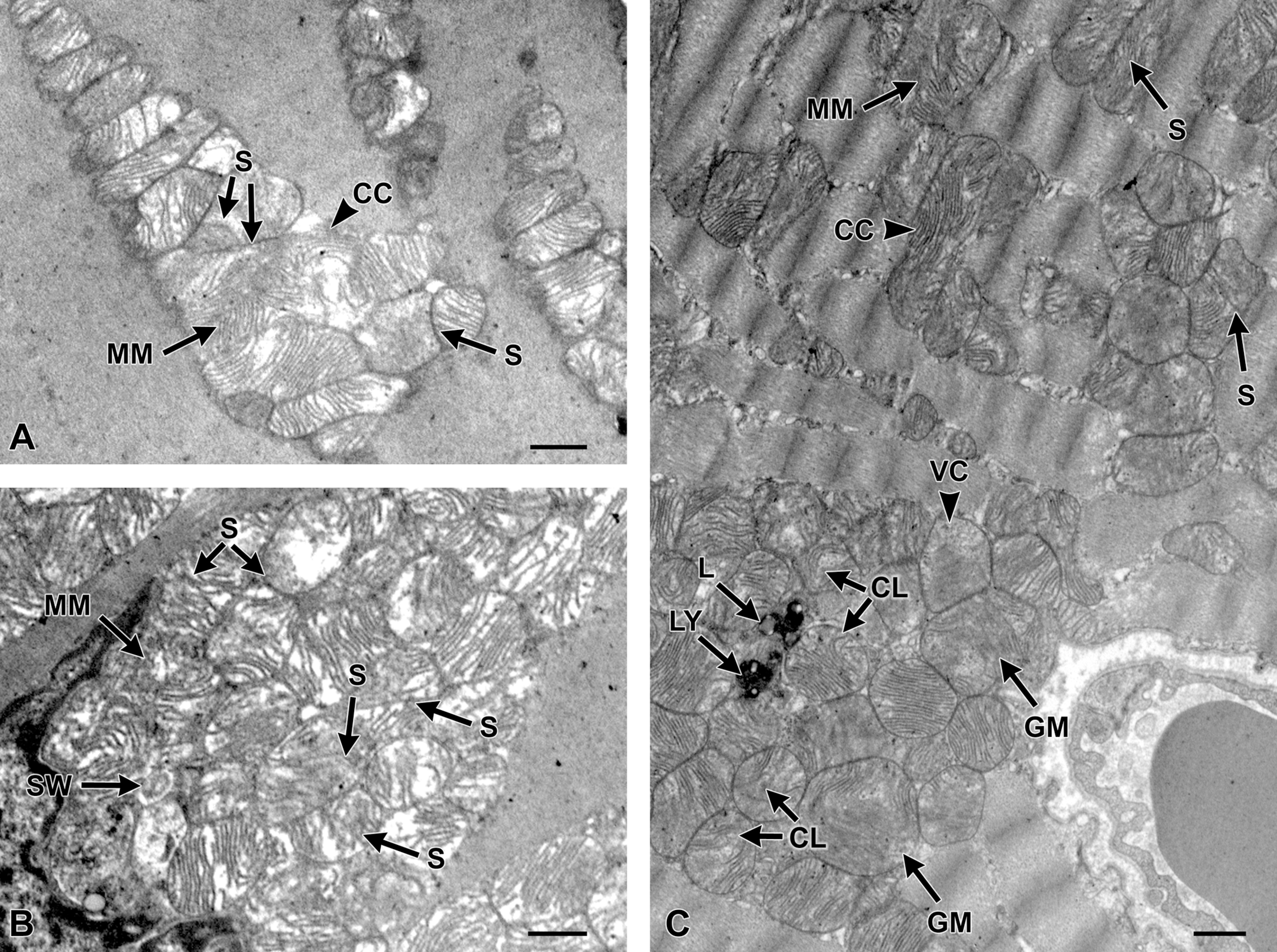
Particle-treated heart sample images (A, B, and C). These particular images further support the findings in the 2.5 mg/kg PM-treated heart samples. In these images, there is marked mitochondria swelling (A and B) characterized by diffuse increasing expansion, fragmentation, swirling [SW], concaving [CC], and looping [CL] of cristae with increased disorganization and vesiculation [VC]. In both images (A) and (B), there was an increase in fission [septation, S] and fusion [megamitochondria, MM; and giant mitochondria, GM] formation. Besides changes to the mitochondria in image (C), there were randomly scattered lysosomes [LY] and lysosomes with lipid droplets [L] not commonly seen in control samples. Bar indicates 2 microns.
Discussion
In real-world conditions, air pollution is inhaled either acutely or chronically (Hassing et al. 2009; Pope et al. 2002, 2006). Herein, we used a model that simulated acute exposure to PM at very high concentration. Health effects caused by PM may vary depending on the composition, concentration, mode, and duration of exposure. In addition to organic substances, PM collected in urban areas in the US contain metals such as zinc (Zn), nickel (Ni), vanadium (V), iron (Fe), manganese (Mn), and copper (Cu). It was previously shown that rats exposed to zinc exhibit broad spectrum of cardiac transcriptional changes, reflective of direct zinc effect on the heart (Gilmour, Schladweiler, et al. 2006). Subsequently it was shown that PM-associated and bioavailable zinc and other metals actually translocate to the circulation and heart, which corroborates with transcriptional changes. (Wallenborn, Kovalcik, et al. 2009; Kodavanti et al. 2008) . Although recent studies have linked PM-associated vanadium and nickel to cardiovascular impairments in humans (Bell et al. 2009) and in ApoE null mice upon long-term inhalation (Chen and Lippmann 2009), the cardiac biochemical and ultrastructural impairments are less well understood. It was recently shown that metals have a damaging effect on cardiac mitochondrial enzymes (Wallenborn, Schladweiler, et al. 2009). Since soluble metals originating from instilled PM could translocate to the heart and other extrapulmonary organs (Wallenborn et al. 2007), we proposed to examine cardiac effects of vanadium-rich PM. In the present study, we found that instillation of high dose of soluble vanadium-rich PM reduces tolerance to I/R, corroborating previous reports (Ballinger 2005; Cozzi et al. 2006; Kang and Hamasaki 2005). The injured myocardium exhibits no abnormalities in mitochondrial dehydrogenase activity compared to the control counterpart under oxygenated conditions. However, under conditions of simulated I/R, exposure to high-dose PM resulted in a decreased mitochondrial dehydrogenase activity. This functional impairment is associated with the ultrastructural alterations in myocardial mitochondria.
The suggested pathways leading to cardiovascular effects of air pollution have been linked to oxidative stress, pulmonary and systemic inflammation, endothelial cell dysfunction, accelerated atherosclerosis, and altered cardiac autonomic function (Brook and Rajagopalan 2010; Pope et al. 2006). Most studies were focused on the impact of PM on arteries, endothelial cells dysfunction, and thrombosis, but only few of these studies on biochemical and molecular alterations in actual myocardial cells (Brook et al. 2004). Exposure of patients with stable coronary heart disease to dilute diesel exhaust caused mild myocardial ischemia and inhibited endogenous fibrinolytic capacity (Mills et al. 2007). Cardiac gene expression pattern was examined in rats exposed to diesel exhaust particles by inhalation over 1 month. Interestingly, there were no findings of pathological or inflammatory changes in the heart in that study. However, this diesel exposure was associated with marked inhibition of mitochondrial aconitase activity (indicative of oxidative stress) and genes involved in cardiac function, antioxidant compensation, and matrix metabolism (Gottipolu et al. 2009).
Because of the tolerance capacity of healthy myocardial tissue, a small degree of cardiac injury might not be apparent as long as the myocardium receives adequate oxygen supply and is not exposed to excess reactive oxygen species. (Ad et al. 2005; Golomb et al. 2009, 2007; Tong et al. 2009). We hypothesized that functional acute and potentially reversible impairment in cardiac mitochondria cannot be detected by light microscopic analysis of the myocardium and that TEM might detect evidence of mitochondrial toxicity following PM exposure. In a previous study, although diesel exhaust particle inhalation was associated with inhibition of mitochondrial aconitase activity, likely reflecting oxidative stress, ultrastructural alterations in the cardiac mitochondria could not be detected (Gottipolu et al. 2009). However, in this study we believed that high-concentration, one-time bolus exposure of rats to vanadium-rich PM would cause ultrastructural changes in myocardial mitochondria. We indeed were able to demonstrate PM-exposure-related changes in mitochondrial integrity. The ultrastructural alterations include mitochondrial swelling, cristae structural deformities, as well as overall abnormalities in the appearance of the sarcoplasm (Ghadially 1997; Griparic and van der Bliek 2001; Meeusen et al. 2006; Rhodin 1975). Mild mitochondrial swelling observed in the control group probably results from the transient hypoxia during the anesthesia and trachea catheterization, and instillation of saline. In rats exposed to HP-10, we find cristae structures and swelling that were often noted in conditions of mitochondrial oxidative stress (Haffor and Alttas 2010). In addition, giant mitochondria and megamitochondria have been known to develop in certain types of cardiomyopathy (Hoppel et al. 2009). Thus, vanadium-rich PM at high concentration causes cardiotoxicity and results in impaired mitochondrial response to I/R.
The effect of PM on myocardial mitochondria and ischemic tolerance was noted 48 hr, but not 24 hr postinstillation. PM has been shown to accumulate in the myocardium already within 4 hr of instillation, where it persists also at 24 hr and 48 hr (Wallenborn et al. 2007).
We believe that the decreased myocardial ischemic tolerance stems from a direct effect of PM. The finding that ischemic tolerance was reduced 48 hr, but not 24 hr, after instillation shows that this effect is not immediate following the myocardial exposure to the particles. On the other hand, the possibility that pulmonary inflammatory mediators contribute, at least partly, to the cardiotoxicity still exists.
Thus, the delay in cardiac mitochondrial impairment in the present study might be due to cumulative effect of existing pulmonary inflammation and associated hypoxia, together with delayed effects of translocated metals.
No studies have provided a link between in vivo PM-exposure, especially in regards to the role of vanadium containing PM, mitochondrial functional impairment, and alteration in ultrastructural integrity of myocardial mitochondria. Our data supports the epidemiological evidence and provides causal relationship between anthropogenic vanadium-rich PM exposure and cardiac impairment.
One of the major limitations of our finding is that the concentration of combustion source PM used in the study was extremely high when compared to long-term inhalation concentrations achieved in polluted areas. The reason we selected relatively high and single instillation doses to study cardiotoxic effect is due to the fact that animal studies addressing the impairment of myocardial mitochondria are relatively few and, furthermore, show negative or subtle effects at ambient concentration, even though the human associations of PM and cardiovascular mortality are robust and consistent. Although cardiac physiological changes have been noted at environmentally relevant concentrations, the data on cardiac biochemical and morphological impact are primarily sparse as the techniques employed are not sensitive to detect subtle mitochondrial or ultrastructural effects. In this study, we elected to use high concentrations to provide a causal link between vanadium-rich PM exposure and cardiac mitochondrial and ultrastructural alterations.
In conclusion, we provide the evidence that single high-concentration pulmonary exposure of rats to soluble vanadium-rich PM causes myocardial mitochondrial functional impairment when subjected to low oxygen/low glucose ischemia. This functional impairment is associated with ultrastructural alterations in mitochondria characterized by cristae shrinkage/breakage and swelling of the inner mitochondrial membrane.
Footnotes
Eliahu Golomb and Didi Matza contributed equally to this work.
This article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency and approved for publication. Approval does not signify that the contents necessarily reflect the views and the policies of the Agency nor does mention of trade names or commercial products constitute endorsement or recommendation for use. This study was supported by the Aaron Beare Foundation, South Africa.