
Abstract
Aberrant signaling by transforming growth factor-β (TGF-β) and its type I (ALK5) receptor has been implicated in a number of human diseases and this pathway is considered a potential target for therapeutic intervention. Transforming growth factor-β signaling via ALK5 plays a critical role during heart development, but the role of ALK5 in the adult heart is poorly understood. In the current study, the preclinical toxicology of ALK5 inhibitors from two different chemistry scaffolds was explored. Ten-week-old female Han Wistar rats received test compounds by the oral route for three to seven days. Both compounds induced histopathologic heart valve lesions characterized by hemorrhage, inflammation, degeneration, and proliferation of valvular interstitial cells. The pathology was observed in all animals, at all doses tested, and occurred in all four heart valves. Immunohistochemical analysis of ALK5 in rat hearts revealed expression in the valves, but not in the myocardium. Compared to control animals, protein levels of ALK5 were unchanged in the heart valves of treated animals. We also observed a physeal dysplasia in the femoro-tibial joint of rats treated with ALK5 inhibitors, a finding consistent with a pharmacological effect described previously with ALK5 inhibitors. Overall, these findings suggest that TGF-β signaling via ALK5 plays a critical role in maintaining heart valve integrity.
Introduction
Transforming growth factor-β (TGF-β) signaling is essential for a wide variety of cellular processes including proliferation, migration apoptosis, and for physiological processes including development, angiogenesis, and immunity (Bertolino et al. 2005; Massagué 2008; Patterson and Padgett 2000; Ten Dijke et al. 2002). TGF-β family of ligands include the TGF-β isoforms (TGF-β1, 2 and 3), activins, nodals, bone morphogenic proteins (BMPs), growth and differentiation factor (GDF), and Mullerian Inhibitory Factor (MIF) (Rahimi and Leof 2007). The TGF-β ligands signal through type I, II, III, and V TGF-β receptors. To date, seven type I, five type II, and one each of the type III and type V members have been described. Transforming growth factor-β ligands preferentially bind to type II serine/threonine kinase receptors, which then bind to and activate type I receptors (activin receptor-like kinases, ALK1–7) in the complex (Rahimi and Leof 2007). ALK5 signaling results in activation of the transcriptional coregulators Smad2 and Smad3, whereas ALK1 or ALK2 activate Smad1, Smad5, and Smad8 (Shi and Massagué 2003). The receptor-associated Smads form homo- and heteromeric complexes involving the common mediator Smad4, which translocate to the nucleus and regulate transcription of target genes (Bierie and Moses 2006).
Deregulation of the TGF-β signaling pathway has been implicated in the pathogenesis of cancer, a range of fibrotic conditions including liver, renal, and pulmonary fibrosis and vascular dysfunctions (De Gouville and Huet 2006; Gauldie et al. 2007; Gordon and Blobe 2008; Goumans et al. 2009; Laping 2003). There is significant current interest in targeting the TGF-β pathway for a range of therapeutic indications, including renal disease (Laping 2003) and advanced stages of cancer (Lahn et al. 2005). The role of TGF-β signaling in cancer is complex and contradictory. In the early stages of carcinogenesis, TGF-β acts as a tumor suppressor by inducing cytostasis and apoptosis, but it switches to a tumor promoter later in carcinogenesis, promoting tumor growth and invasion, evasion of immune surveillance, and cancer cell dissemination (Bierie and Moses 2006; Massagué 2008). In addition, reduced TGF-β signaling has been implicated in compromised wound healing and high levels are associated with excessive scarring (Roberts and Sporn 1993).
The TGF-β superfamily signaling pathways play a key role during cardiac development (Sridurongrit et al. 2008), which was first suggested when TGF-β2−/− embryos were found to display multiple cardiac defects including ventricular septum defects and myocardial thinning (Sanford et al. 1997). Specific loss of ALK3/BMPR1a in the myocardium of atrioventricular (AV) cushions results in valve malformations, whereas deletion in the endothelium reduces epithelial-mesenchymal transition (EMT) of the AV cushion, resulting in defective cardiac valve and septa formation (Gaussin et al. 2005; Song et al. 2007). A role for ALK2 in cardiac development has also been reported. Chick AV cushion explants fail to undergo EMT when exposed to ALK2-neutralizing antibodies, and endothelium-specific deletion of ALK2 in mice also results in a loss of EMT (Lai et al. 2000; Wang et al. 2005). Furthermore, congenital heart defects are suspected in individuals carrying mutations in the ALK2 gene (Smith et al. 2009).
A number of small molecules that inhibit ALK5 have been developed and demonstrate encouraging results in animal models of renal fibrosis (Gellibert et al. 2006) and cancer (Li et al. 2008; Suzuki et al. 2007; Uhl et al. 2004). However, questions remain over the homeostatic role of ALK5 signaling, and therefore the safety implications of targeting this molecule. ALK5 is required for the induction of EMT during heart development in the chick (Mercado-Pimentel et al. 2007; Sridurongrit et al. 2008), and mice embryos lacking ALK5 die around E10.5 because of aberrant vasculogenesis in the yolk sac (Larsson et al. 2001). ALK5-null embryos also exhibit a defect in the formation of vascular smooth muscle layers (Seki et al. 2006). ALK5 does not play a role in cardiomyocyte development in the mouse, but as in the chick, it is important in the endocardium for induction of EMT both in vitro and in vivo (Sridurongrit et al. 2008). Although a role of TGF-β signaling in cardiac development has been established, the role of ALK5 in maintaining cardiac homeostasis is poorly understood and the cardiovascular effect of inhibiting this kinase in adult mammals is unknown.
We have recently developed chemical inhibitors of ALK5. The compounds are derived from two independent chemical series; one of the series has been published recently (Goldberg et al. 2009). While performing preclinical toxicity studies with these compounds in rats, we observed very unusual histopathologic heart valve changes. The lesions were induced by ALK5 inhibitors from two different chemistry series, indicating that the pathology occurred as a consequence of ALK5 inhibition rather than via an off-target effect. Interestingly, immunohistochemical analysis revealed that in the heart, ALK5 expression was unique to the valves. This study has highlighted the difficulties associated with therapeutically targeting the ALK5 pathway and identified a role for ALK5 in maintaining heart valve integrity in rats.
Materials and Methods
Test Compounds
The two test compounds AZ12601011 and AZ12799734 were synthesized by AstraZeneca Pharmaceuticals, Cheshire, UK. In vivo dosing suspensions were prepared in 0.5% w/v HPMC/0.1% w/v Tween 80.
ALK5 Inhibitor In Vitro Selectivity Screening
Selectivity testing was accomplished using a KinomeScan competition binding assay (KinomeScan, DiscoveRx, San Diego, CA, USA), which measures the ability of a compound to compete with an immobilized active site probe molecule. Individual DNA-tagged kinases, expressed by phage display in E.coli, were used in an affinity capture assay on beads coated with immobilized active site ligand. After washing excess unbound kinase, qPCR targeting the DNA tag is used to determine levels of bound kinase (Fabian et al. 2005). In this case, test compound binding affinity was determined using an eleven-point dose response in the range 0.5 nM–30,000 nM.
Smad2 Redistribution Assay
The Smad2 redistribution assay (ThermoFisher Scientific, Runcorn, UK) used MDA-MB-468 cells stably expressing human Smad2 fused to the C-terminus of enhanced green fluorescent protein (EGFP). This assay allows evaluation of inhibitors of TGF-β1 induced Smad2 translocation from the cytoplasm to the nucleus, a process that requires ALK5 activity (Liu et al. 2009). The assay was performed according to the manufacturer’s instructions. Briefly, the test compound and 2 ng/mL TGF-β1 (Calbiochem-Novabiochem, Nottingham, UK) were added to pre-plated MDA-MB-468 cells and incubated for ninety minutes. Cells were then fixed, stained with Hoechst 33342 (1 µM) and high-content imaging using a ThermoFisher Cellomics Arrayscan VTI reader was performed. Briefly, plates were read using 10 × 0.3 NA objective and an XF53 filter set. Data analysis was performed using the compartmental analysis V2 bioapplication, in which the ratio of cytoplasmic to nuclear intensity was expressed on a per cell basis. Percentage inhibitory activity for test compounds at various concentrations was calculated relative to a positive control antagonist SB431542 used at 3 µM (Sigma-Aldrich, Poole, UK) and a negative control (0.25% DMSO) and an IC50 value estimated using Origin (Originlab Corp, Northampton, MA, USA).
In Vivo Studies
The animals used in this study were ten-week-old female HsdHan:WIST rats obtained from Harlan, UK. Studies were carried out in accordance with UK Home Office legislation (Animals [Scientific Procedures] Act 1986) and AstraZeneca’s institutional policies. In an initial experiment, AZ12601011 at doses of 200–300 mg/kg/day or AZ12799734 at doses of 100–400 mg/kg/day were orally administered for three days with necropsy twenty-four hours after the final dose (two animals per compound per dose). In the main phase of the study, AZ12601011, AZ12799734, or vehicle control was orally administered once daily for up to seven days. AZ12601011 was dosed at 150 or 300 mg/kg/day, and AZ12799734 was dosed at 200 or 400 mg/kg/day with four animals per compound per dose. Water and food were available ad libitum. Animals were killed for necropsy by administration of halothane.
Pathology
At necropsy, external features were inspected and the thoracic and abdominal cavities and contents were examined. As much blood as possible was withdrawn from the V. cava, in order to empty the heart chambers. Macroscopic abnormalities were recorded, and hearts and femoro-tibial joint were sampled, fixed in 10% buffered formalin, and processed. The hearts were embedded longitudinally in paraffin, and step sections at nominal thickness of 5 µm were cut every 100 µm to identify all four heart valves. Sections were stained with hematoxylin and eosin (H&E) and examined microscopically. Femoro-tibial joints were decalcified prior to sectioning and H&E staining.
ALK5 Immunohistochemistry
For immunohistochemistry, sections were dewaxed in two changes of xylene, rehydrated through two changes of 100% and 95% alcohol, and placed into running tap water. Endogenous peroxidase activity was blocked with 3% (aq) hydrogen peroxide for ten minutes, after which sections were washed in tap water. Sections were placed in a pressure vessel containing 0.01 M citrate buffer pH 6.0, and antigen retrieval was performed using Milestone’s Rapid Microwave Histoprocessor RHS-2 (Milestone, Sorisole, Italy). Tissue sections were heated to 110°C and maintained at this temperature for two minutes. The vessel was then cooled for ten minutes using running tap water, and sections were then removed and rinsed in tap water for five minutes. Sections were transferred to a Lab Vision autostainer (Thermo Fisher Scientific, Runcorn, UK) for IHC. Following a wash in Tris-buffered saline with 0.05% Tween (TBST at pH 7.4), sections were protein blocked by application of normal goat serum (DAKO, X0907) diluted to 1:20 in TBST, for twenty minutes. Sections were incubated for sixty minutes at room temperature with an affinity-purified rabbit polyclonal antibody to ALK5 (ab31013 Lot: 327199, Abcam, Cambridge, UK), diluted to 1:30 in TBST. Further heart sections were included as negative control, omitting the primary antibody. Previous work in our laboratory, using rabbit IgG isotype (Dako, Ely, UK) as a negative control, had confirmed the specificity of this antibody in rat hearts and primate placenta.
Results
ALK5 Inhibitor Kinase Selectivity Profiles
Two compounds (AZ12601011 and AZ12799734) were identified as potent inhibitors of ALK5 in both an in vitro competition binding assay and a cell-based ALK5 inhibition assay. Data derived from these assays for AZ12601011 and AZ12799734 are shown in Table 1. In addition to being potent against ALK5 in the binding assay (inhibitory constant [Kd’s] 0.0029 and 0.74 µM, respectively) both compounds were also highly active in cellulo ALK5 inhibitors (IC50 0.022 and 0.017), as measured by the ability of the compounds to inhibit nuclear translocation of Smad2 in MDA-MB-468 cells. It is important to note that in addition to inhibiting ALK5, both compounds also exhibited in vitro inhibitory activity against the closely related ALK family members ALK4 and ALK6 (Table 1). AZ12601011 inhibited ALK4 with a Kd of 0.0026 µM and ALK6 with a Kd of 0.042 µM, and AZ12799734 inhibited ALK4 with a Kd of 1 µM and ALK6 with a Kd of 0.017 µM. Both compounds showed weak activity against ALK1, ALK2, and ALK3 (Table 1).
ALK5 inhibitor selectivity profiles. Kinase inhibition data of both compounds of different ALK family members and cellular inhibition of ALK5.
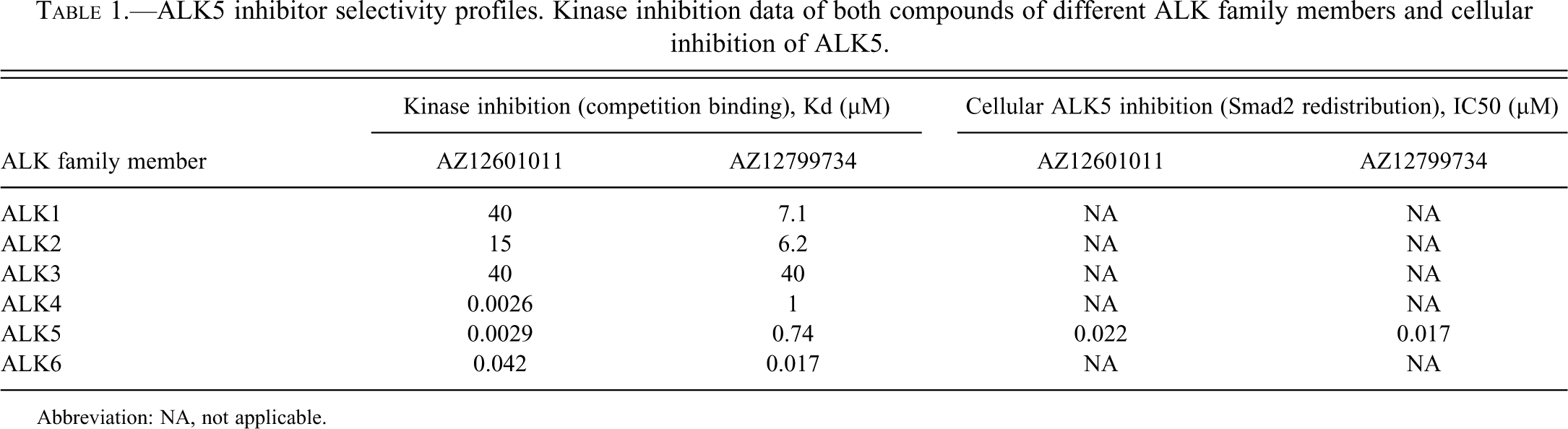
Abbreviation: NA, not applicable.
Heart Valve Lesions in ALK5 Inhibitor-Treated Animals
There were no significant macroscopic findings in any of the hearts on external examination. Microscopic evaluation revealed heart valve lesions in response to treatment with either AZ12601011 or AZ12799734. Lesions were not restricted to one particular heart valve and were detected in all four valves in the majority of treated animals. There was no apparent dose dependency with respect to generation of the heart valve pathology, as it was observed at all doses tested in our studies. Heart valve lesions consisted of hemorrhage, inflammation, degeneration, and valvular interstitial cell activation and proliferation.
No microscopic changes were present in any heart valve of the control animals, as assessed by comparable numbers of step sections (Figure 1A and 1B). In ALK5 inhibitor–dosed rats, hemorrhage into the heart valves was evident at low magnification (Figure 1C). The normal architecture of the leaflet was replaced by hemorrhage (Figure 1D).
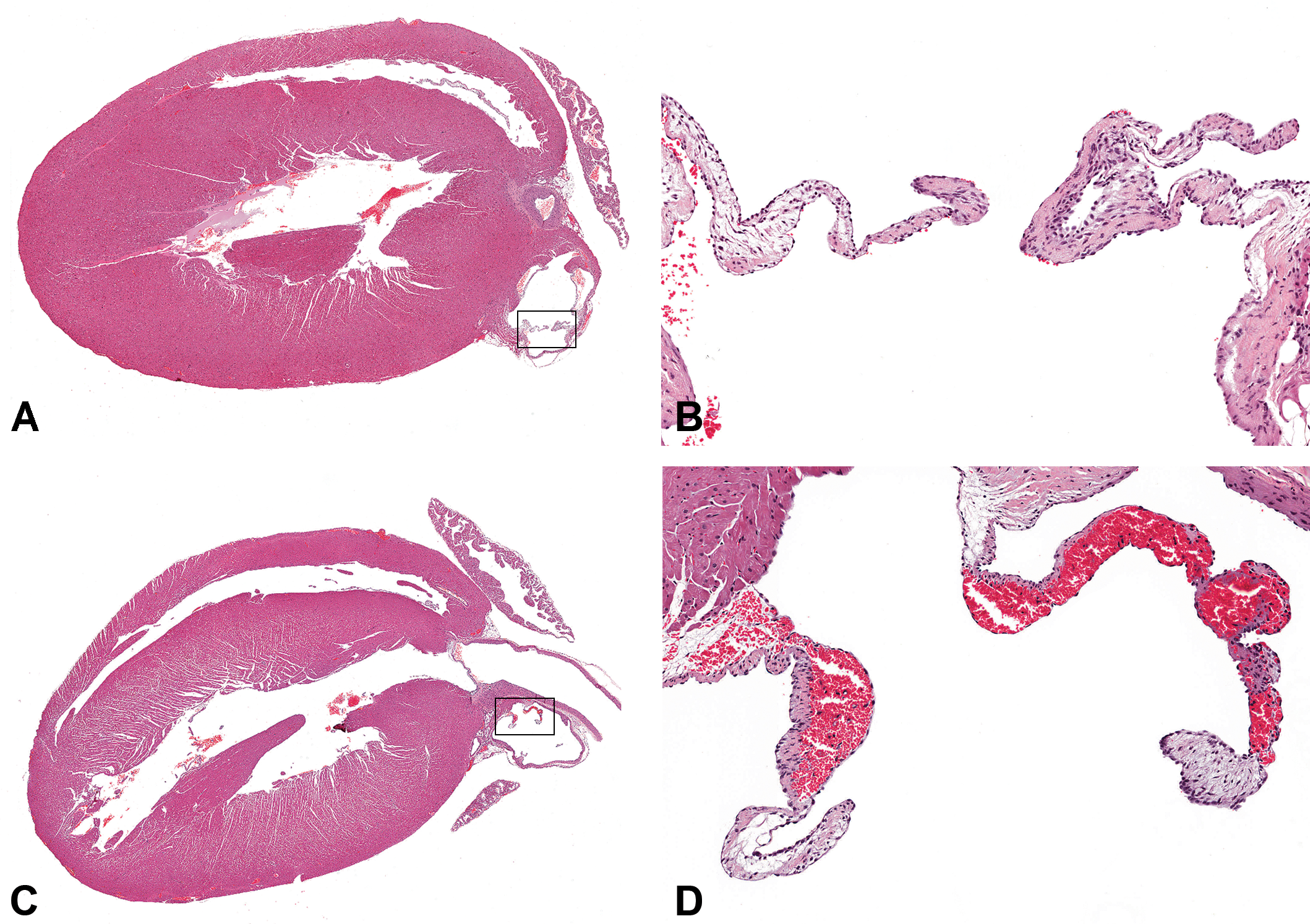
Comparison of heart valves in control and ALK5 inhibitor–treated rats, hematoxylin and eosin stain. (A) Overview of longitudinal section of control heart; ×7. The rectangle contains a leaflet of the pulmonary artery valve, shown at higher magnification in B. (B) Higher magnification of pulmonary valve of control rat; ×100. (C) Overview of longitudinal section of heart from rat dosed with 200 mg/kg/day of AZ12799734 for five days; ×7. Note the leaflet of the pulmonary artery with hemorrhage (within rectangle, shown at higher magnification in D). (D) Higher magnification of valve cusp of the pulmonary artery from rat dosed with 200 mg/kg/day of AZ12799734 for five days; ×100. The heart valve is enlarged by severe hemorrhage.
After dosing of either ALK5 inhibitor for three days, the main histopathologic change consisted of minimal to mild focal hemorrhage, often present in individual step sections in only one or two valves. In some animals, minimal focal infiltration of neutrophils into the valve leaflet was noted. A minimal focal increase of valvular interstitial cells, which represent the main cellular component of the heart valves, was seen in single step sections of some of the animals dosed for three days.
After dosing ALK5 inhibitors for five to seven days, the heart valve lesions were more extensive and present in the majority of step sections through the heart. The activation and proliferation of valvular interstitial cells had progressed and was noted in all animals in at least one valve. In control animals, the valvular interstitial cells were elongated, with inconspicuous cytoplasm and a small, elongated nucleus (Figure 2A). After treatment with either ALK5 inhibitor, valvular interstitial cells were found to be increased in size and number, with increased cytoplasm, an enlarged round to spindeloid nucleus, and frequently undergoing mitosis (Figure 2B). In contrast, mitoses of valvular interstitial were not seen in control animals of the same age. In numerous heart valves of ALK5 inhibitor-dosed animals, signs of degeneration were present; these signs were characterized by presence of cellular debris and fibrin, often accompanied by infiltration of inflammatory cells, mainly neutrophils (Figure 2B). Compared to dosing for three days, hemorrhage was more extensive after five to seven daily doses and was usually noted in three or all four heart valves. The lesion had also progressed from presence of focal erythrocytes in a single step section to severe hemorrhage expanding the valve leaflet over several adjacent step sections in one given heart valve (Figure 2C). Inflammatory cell infiltration and proliferation of valvular interstitial cells was more severe and present in a higher proportion of valves after five to seven days of dosing (Figure 2D).
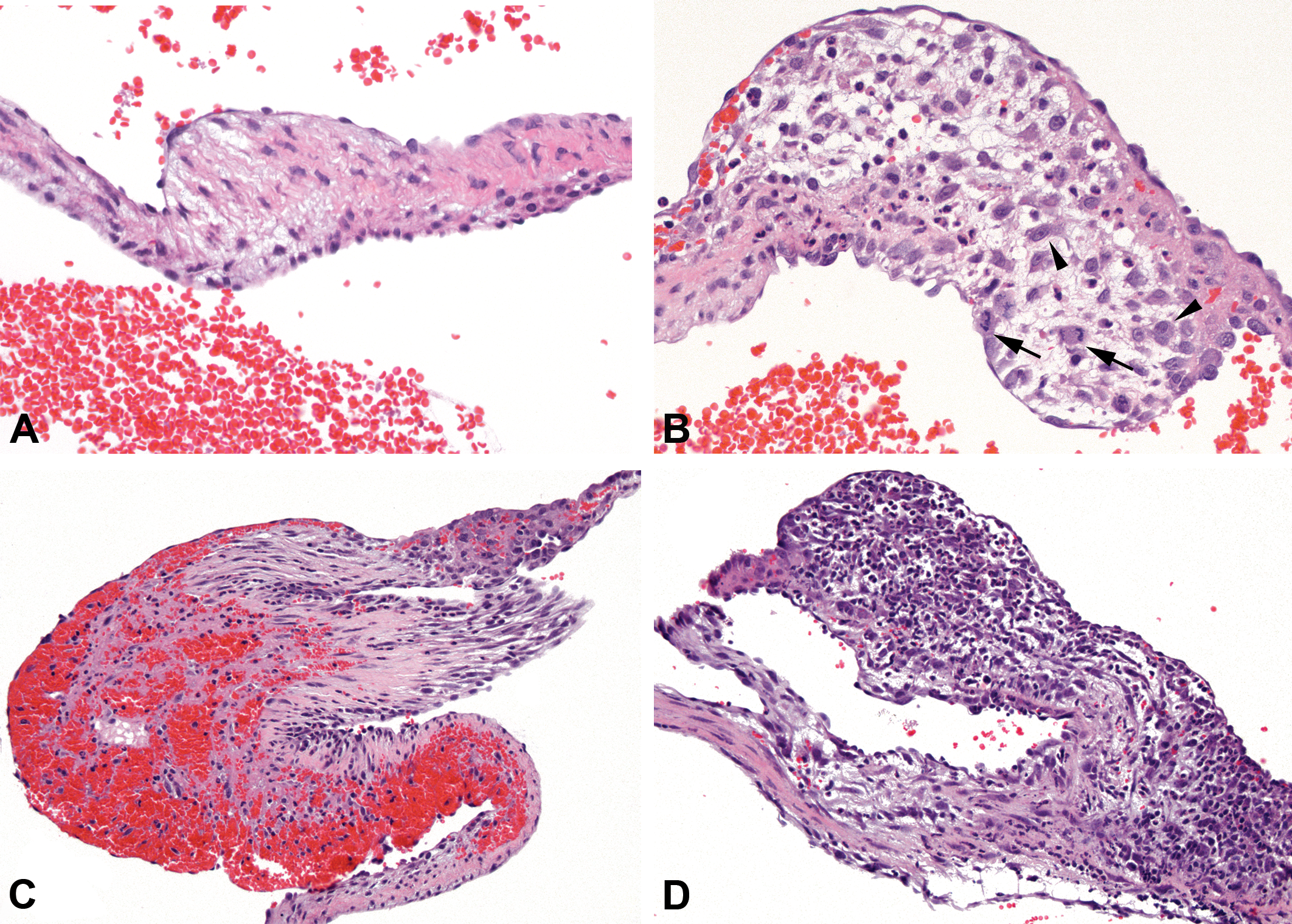
Comparison of heart valves in control and ALK5 inhibitor–treated rats, hematoxylin and eosin stain. (A) Aortic heart valve of a control rat. The valvular interstitial cells are small with inconspicuous cytoplasm; ×400. (B) Aortic valve from rat dosed with 200 mg/kg/day of AZ12799734 for five days, taken at comparable anatomic location as A. Valvular interstitial cells are enlarged (black arrowheads) and increased in numbers displaying several mitoses (black arrows). Degenerated cells, neutrophils, and hemorrhage are present; ×400. (C) Rat dosed with 400 mg/kg/day of AZ12799734 for six days. The leaflet of the right atrioventricular valve is severely expanded by hemorrhage; ×200. (D) Aortic valve from rat dosed with 150 mg/kg/day of AZ12601011 for seven days. Severe infiltration of mainly neutrophilic granulocytes, fewer macrophages and lymphocytes, along with proliferation of valvular interstitial cells; ×200.
Next, we hypothesized that the valvular lesions, which developed upon treatment of rats with ALK5 inhibitors, arose as a consequence of specific blockade of ALK5 signaling in the valve. To explore this hypothesis, we performed immunohistochemistry for ALK5 on the hearts, using a rabbit polyclonal antibody raised against a synthetic peptide representing amino acids 158–179 of ALK5. The heart valve (Figure 3A and 3B), but not the myocardium (Figure 3C) or other cardiac structures, stained positively for ALK5 expression. ALK5 was expressed focally in the heart valve, predominantly along the endocardium, in a linear manner, consistent with a membranous localization (Figure 3A and 3B). The distribution of positive ALK5 staining varied from a single positive focus to multifocal positive staining per individual heart valve. In ALK5 inhibitor–treated rats, occurrence of positive ALK5 staining was unrelated to histopathologic lesions. No difference in distribution or intensity of valvular ALK5 expression was observed between controls or animals dosed with ALK5 inhibitors for up to seven days. Overall, these data indicate that in the rat heart, ALK5 is specifically expressed in the valves.
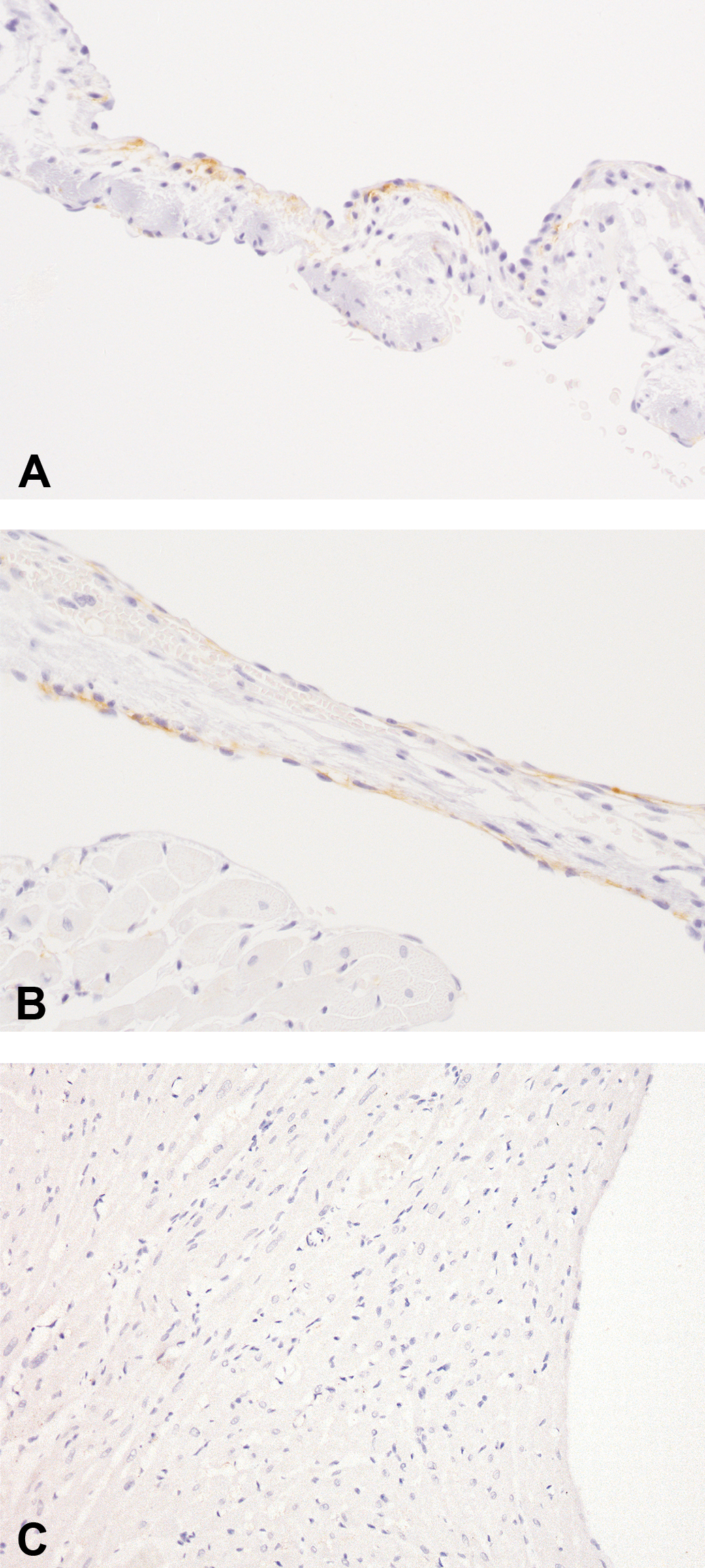
Comparison of ALK5 expression in the heart valve and myocardium. Immunohistochemical detection of ALK5 expression in the heart. (A) Right atrioventricular valve from a control rat. Positive staining for ALK5 is present focally beneath the endocardial lining; ×400. (B) Right atrioventricular valve from rat dosed with 150 mg/kg/day of AZ12601011 for seven days; ×400. Staining for ALK5 in this valve with hemorrhage is comparable to staining in the control animal. (C) Negative myocardium; ×200.
Physeal Dysplasia in ALK5 Inhibitor–Treated Animals
Physeal dysplasia has been described previously in toxicity studies evaluating the effects of ALK5 inhibitors (Frazier et al. 2007). Therefore, we decided to examine the femur and tibia of animals treated with AZ12601011 and AZ12799734 to evaluate whether dysplasia was also a property of our ALK5 inhibitors. An increase in femur and tibia growth plate thickness compared to control animals (Figure 4A) was observed after treatment of rats with AZ12799734 for six days (Figure 4B) or AZ12601011 for seven days (Figure 4C). Primarily the hypertrophic zone, and to a lesser extent the proliferative zone, of the growth plate were affected by an increase in thickness. Chondrocyte columns were irregularly arranged, and individual chondrocytes in the hypertrophic zone were enlarged, displaying swollen cytoplasm. Occasional foci of disruption of the growth plate, accompanied by mild hemorrhage, were present in the metaphysis.
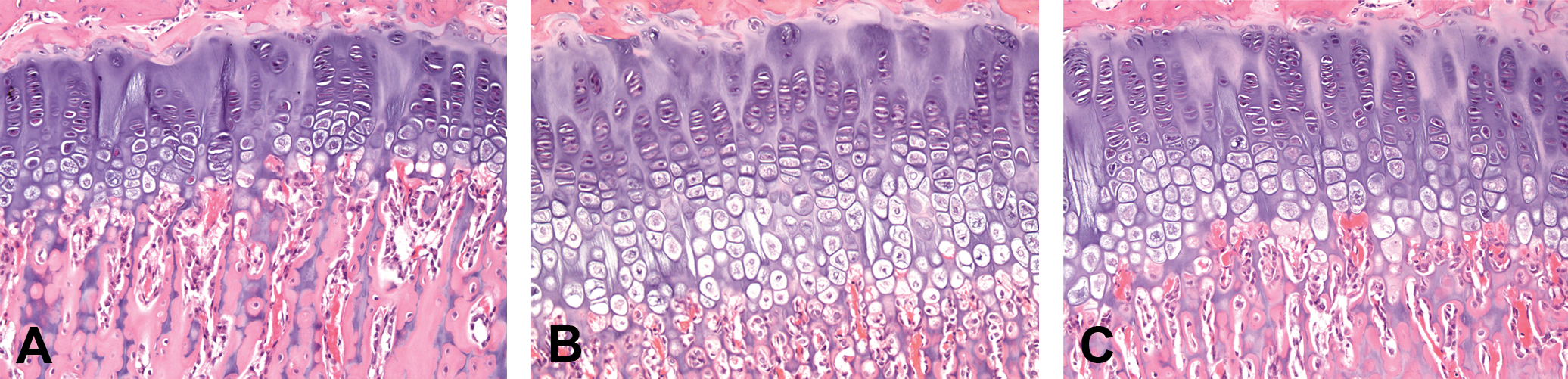
Physeal dysplasia of the tibia in ALK5 inhibitor–treated rats. Hematoxylin and eosin–stained section of growth plate of the tibia, all ×200. (A) Control animal. (B) Rat treated with 400 mg/kg/day of AZ12799734 for six days. (C) Rat treated with 150 mg/kg/day of AZ12601011 for seven days. Thickening of the hypertrophic and proliferative zone of the growth plate in ALK5 inhibitor–treated animals.
Discussion
In this study, treatment of rats with ALK5 inhibitors was associated with the development of microscopic heart valve lesions. The incidence and severity of the lesions described here support a direct ALK5 inhibitor–mediated effect. The lesions were of degenerative nature with hemorrhage and inflammation and of reactive nature with activation and proliferation of valvular interstitial cells, the most prevalent cells of the heart valves.
Given the promiscuous nature of kinase inhibitors and the homology between ALK family members, it could be argued that the valvular lesion is associated with another ALK family member. Both compounds were screened against ALK1-6. Both compounds have low affinity for ALK1, ALK2, and ALK3 but notably retained considerable activity against ALK4 and ALK6. However, it is important to recognize that ALK4 is not expressed in the heart (Ten Dijke et al. 1993) and ALK6 KO mice do not have cardiovascular abnormalities (Yi et al. 2000), which add further support to the theory that the valvular lesions reported here were attributable to inhibition of ALK5 signaling rather than inhibition of other ALK family members. ALK5 expression restricted to the heart valves could be demonstrated and further endorses a direct target effect. However, using immunohistochemistry, no difference in valvular ALK5 expression was evident between treated and control animals. Nevertheless, a differentiation between the inactive and active forms of ALK5 was not possible with the commercial antibody used, and therefore the amount and intensity of ALK5 staining does not reflect activity of ALK5.
In the present study, valvular pathology was observed at doses that also caused a physeal dysplasia. Recently, inhibitors of ALK5 were reported to induce physeal dysplasia in rats with expansion of the hypertrophic and proliferative zones (Frazier et al. 2007). Physeal lesions have been previously ascribed to disruption of chondrocyte maturation, which is regulated by multiple interacting kinase pathways, including TGF-β/ALK5. In addition, TGF-β receptor KO mice have physeal and limb abnormalities (Serra et al. 1997). Taken together, these results provide further evidence that the lesion occurs via an ALK5-mediated pharmacological mechanism, since the lesion to the heart valves occurs at doses that produce other known ALK5-mediated pharmacology.
Drug-induced valvular disease has been described for agents used to treat obesity (fenfluramine and phentermine), Parkinson’s disease (ergot derivatives pergolide and cabergoline), and migraine (ergotamine and methysergide) (Connolly et al. 1997; Hauck et al. 1990; Schade et al. 2007; Zanettini et al. 2007). There is increasing evidence that 5-hydroxytryptamine 2B receptor (5HT2BR) activation and/or increased circulating 5-hydroxytryptamine (5-HT) may play a significant role in drug-induced valvulopathy (Elangbam et al. 2008; Fitzgerald et al. 2000). Compounds in this study were screened against 5HT1A, 5HT2A, 5HT2B, 5HT3, and 5HT4 receptors and found to be inactive at a concentration of 10 µM (<5% inhibition; data not shown). These data indicate the pathology observed with ALK5 inhibitors is independent of 5HT receptor activity. The finding that the lesion was induced by ALK5 inhibitors from two different chemistry series indicates that the pathology most likely occurred as a consequence of ALK5 inhibition rather than via an off-target effect.
Pathologic conditions of the heart valve can cause hemodynamic overload to the left or right, or both ventricles, which eventually leads to myocardial dysfunction and heart failure and sometimes to sudden death (Carabello and Crawford 1997). TGF-β1 and its downstream pathways play a critical role in extracellular matrix (ECM) remodeling, tissue repair, and wound healing (Gordon and Blobe 2008; Verrecchia and Mauviel 2001). Based on the known role of TGF-β signaling in tissue repair and wound healing, we hypothesize that by inhibiting ALK5, the normal tissue homeostasis and repair function orchestrated by the TGF-β pathway is abrogated. The heart valves may be particularly affected as a result of ongoing mechanical stress at opening and closure during each heart cycle. The valvular interstitial cells, the most abundant cells of the heart valves, have characteristics of fibroblasts but are a phenotypically dynamic cell population. They maintain the integrity and stability of normal valves and regulate repair processes and produce the different components of the ECM such as collagen, elastin, and glycosaminoglycans. Cellular and noncellular elements of the heart valve accommodate the tissue dynamics across the cardiac cycle. Collagen is the major stress-bearing component to withstand tensile forces, elastin provides flexibility of the heart valve, and glysosaminoglycans absorb shock and shear (Donnelly 2008; Elangbam 2010; Mulholland and Gotlieb 1996; Schoen 2008). Valvular interstitial cells mediate the matrix remodeling and continuously repair damage to collagen in heart valves (Schoen 2008). The ALK5 pathway is generally responsible for TGF-β signaling in fibroblasts, and ALK5 inhibition interferes with ECM production and thus fibrosis (Grygielko et al. 2005; Leask 2010). In response to injury, the valvular interstitial cells may transform from their quiescent state to an activated, myofibroblast-type cell type, expressing increased amounts of α-smooth muscle actin (α-SMA) (Elangbam 2010; Liu and Gotlieb 2008). TGF-β1 has been shown to mediate increased levels of α-SMA in valvular interstitial cells in vivo and is thought to have an important role in the remodeling of the adult heart valve (Walker et al. 2004). Using gene expression profiling in fibroblast cell lines, it could be demonstrated that TGF-β1 signals exclusively through receptor complexes involving ALK5 (Karlsson et al. 2005). When performing immunohistochemistry for α-SMA on hearts with ALK5 inhibitor–induced valve lesions during an earlier study, no difference in α-SMA expression was found in comparison to control animals (unpublished data). Given the key role of TGF-β in maintaining the ECM and the continuous requirement of ECM remodeling in heart valves, the consequence of ALK5 inhibition may be a reduction in valvular strength, resulting in hemorrhage and a secondary inflammatory response. Further investigation into the effect of ALK5 inhibition on the molecular changes of ECM composition and organization within the heart valves is needed.
In routine histological assessment of the heart in preclinical studies, assessment of heart valves is usually performed on a single longitudinal heart section. The sections may contain one or more, but occasionally none of the four heart valves (Elangbam 2010). A study reviewing heart sections from a chronic rat study showed that none of the four heart valves was consistently present in heart sections of 300 rats (Elangbam et al. 2006). Considering the key role of heart valves in cardiac function, the value of systematic histological evaluation of the heart valves during preclinical toxicology studies cannot be underestimated. In this study, a comprehensive evaluation of the heart valves was performed by producing heart step sections in order to assess all four heart valves in each animal.
In summary, these findings suggest a homeostatic role of ALK5 in maintaining heart valve integrity, which is essential for normal cardiac function. This work clearly brings into question the feasibility of systemic modulation of the ALK5 pathway for the safe treatment of disease.
Footnotes
Acknowledgments
We would like to thank Tony Casey, Melanie Marsden, and Jane Reeves for excellent technical assistance.