
Abstract
Several multikinase angiogenesis inhibitors demonstrate mitochondrial and/or cardiovascular toxicity, suggesting an on-target pharmacologic effect. To evaluate whether cardiotoxicity is directly related to vascular endothelial growth factor receptor inhibition, we investigated the effects of sunitinib, sorafenib, and pazopanib on myocardial function and structure. We used a rat model to assess myocardial effects of the inhibitors concurrently exposed to the cardiac stressor dobutamine. Echocardiographic abnormalities including premature ventricular contractions, decreases in heart rate, circumferential strain, and radial and circumferential strain rates were noted with sorafenib, but not with sunitinib or pazopanib. Ultrastructural analysis of ventricular cardiomyocytes by transmission electron microscopy revealed mitochondrial swelling, dense deposits, and matrix cavitation in rats given sunitinib and disrupted mitochondrial cristae in rats given sorafenib, but there were no effects with pazopanib. Effects on neonatal rat cardiomyocyte cultures were assessed, which identified decreases in mitochondrial membrane potential with sunitinib treatment, but not with sorafenib or pazopanib. Intracellular adenosine triphosphate depletion was observed with sunitinib and sorafenib, but not pazopanib. Our results show that cardiotoxicity is not necessarily related to a pharmacologic classwide effect of vascular endothelial growth factor receptor inhibition, and the rat myocardial structural and functional changes identified in this study may be instead a result of inhibition of other kinase pathways, the mechanism of which may be associated with mitochondrial toxicity.
Introduction
Angiogenesis plays a critical role in numerous cancers. Thus, a strong rationale exists for using antiangiogenic agents to target tumor types that express vascular endothelial growth factor receptor (VEGFr), platelet-derived growth factor receptor (PDGFr), or c-kit (Kim and Kaelin, Jr. 2006; Rini 2007). There are several angiogenesis inhibitors, including sorafenib, sunitinib, bevacizumab, and most recently, pazopanib, that have received regulatory approval for cancer therapy. Sorafenib is a Raf kinase inhibitor that inhibits VEGFr-1,-2, -3; PDGFr-β; Fms-like tyrosine kinase-3 (Flt-3); and c-kit. Sunitinib inhibits VEGFr-1, -2, and -3; PDGFr-α and –β; c-kit; Flt-3; colony-stimulating factor Type 1; and the glial cell line derived neurotrophic factor receptor (RET). Bevacizumab, a humanized monoclonal antibody directed against VEGF, is used in combination with interferon. Pazopanib is a potent, multi-targeted tyrosine kinase inhibitor of VEGFr-1, -2, and -3; PDGFr-α and –β; and c-kit, which has demonstrated promising clinical findings (Harris et al. 2008; Hurwitz et al. 2009; Sloan and Scheinfeld 2008). These drugs are marketed for treatment of renal cell carcinoma (RCC) in the United States, Europe, and Australia, with the exception of bevacizumab, which is not licensed for RCC in the United States, and pazopanib, which is licensed in the United States and has gained conditional approval in Europe.
Even though pazopanib, sunitinib, and sorafenib have apparently similar molecular target profiles of VEGF receptors, PDGF receptors, and c-kit, relative affinities among these receptors, as well as affinities for other kinases and other unidentified factors may result in differing efficacy and, especially, toxicity profiles among agents, and they may provide treatment options for patients with RCC.
Although great strides have been made with tyrosine kinase inhibitors and cancer therapy, several have been associated with clinical cardiotoxicity, manifesting as symptomatic congestive heart failure or asymptomatic left ventricle (LV) dysfunction. Specifically, sunitinib (Demetri et al. 2006; Pfizer 2007) and sorafenib (Bayer Pharmaceuticals 2007), approved orally active TKIs, have been associated with cardiotoxicity. These findings, in addition to associations between adenoviral-mediated disruption of VEGFr signaling and increased contractile dysfunction, fibrosis, and heart failure (Izumiya et al. 2006; Shiojima et al. 2005; Wyatt et al. 1980), has led to speculation that cardiotoxicity is a classwide effect of angiogenic (VEGFr) inhibitors. To evaluate this hypothesis, we explored the effects of pazopanib, sunitinib, and sorafenib in a rat dobutamine-induced cardiac stress model and a rat cardiomyocyte in vitro model. We identified compound-selective effects both in vivo and in vitro, indicating that the expression of cardiotoxicity, and potentially even the mechanism of these effects, differ among agents in this class, and we suggest that myocardial and mitochondrial toxicity may not be related to VEGFr inhibition, but rather to other structure activity relationships or to other kinase activity with this class of agents.
Materials and Methods
Compounds, Dose Preparation, and Administration
Sunitinib (malate salt) and sorafenib (p-toluenesulfonate salt) were supplied by LC Laboratories (Woburn, MA), and sorafenib (free base) was supplied by ChemieTek (Indianapolis, IN). Pazopanib (GW786034B) was supplied by the Department of Pharmaceutical Development, GlaxoSmithKline. Doxorubicin and carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) were purchased from Sigma-Aldrich (St. Louis, MO). All test articles were of ≥ 99% purity.
Dosing preparations for sunitinib (1 mg/mL in water), sorafenib (1 mg/mL in 15.4% pluronic F68/42.3% polyethylene glycol 400/ 42.3% propylene glycol), or pazopanib (30 mg/mL in 0.5% hydroxypropylmethylcellulose [K15]/0.1% Tween-80, pH 1.3) were given orally by gavage at a dose volume of 10 mL/kg. A dobutamine (12.5 mg/mL, Hospira, Lake Forest, IL) injection, which was diluted with 0.9% sodium chloride to 125 μg/mL, was given intravenously at a dose of 20 μg/kg/min at an infusion rate of 0.16 mL/kg/min for ten minutes. The total volume given to each rat was based on daily body weight data.
For in vitro studies, except for doxorubicin, which was prepared in phosphate-buffered saline (PBS), all compounds were dissolved in dimethyl sulfoxide (DMSO) and treated at final concentrations of 0.1% (sunitinib, sorafenib, and FCCP) or 0.3% (pazopanib) DMSO.
Animals and Maintenance
Male Crl:CD (SD) Virus Antibody Free (VAF/Plus) rats (Charles River Laboratories, Raleigh, NC, ten to eleven weeks of age) were used in this study. All rats were acclimated to housing conditions, which included environmental enrichment, and the dosing procedure prior to the initiation of dosing.
Rats were housed individually in polycarbonate cages in an environmentally controlled room (64–79°F; 30–70% relative humidity) with a twelve-hour light/dark cycle. All animals were offered approximately 23 g of a laboratory diet (Extruded Certified Rodent Chow 5L35, PMI Nutrition International, Richmond, IN) once daily and water ad libitum except during periods of echocardiographic measurements (up to two hours). All procedures involving animal care and use in this manuscript were conducted in accordance with published guidelines (the National Research Council’s Guide for the Care and Use of Laboratory Animals) and the US Department of Agriculture’s Animal Welfare Act, and they have been reviewed and approved by GlaxoSmithKline’s Institutional Animal Care and Use Committee (IACUC).
In Vivo Study Design
The choice of twenty-one days of duration of treatment and the inclusion of strain and strain rate were based on the results of a parallel echocardiographic study in the rat in which we used doxorubicin administration as a positive control for cardiac toxicity (Burdick et al. submitted for publication). Briefly, doxorubicin (2 mg/kg intraperitoneal weekly) resulted in significantly lower dobutamine-induced increases in radial strain (days 21 and 28) and circumferential strain (day 28).
In a previous four-week, repeat-dose oral toxicity study in the rat, a dose of 300 mg/kg pazopanib was maximally tolerated, and the exposure achieved at this dose was comparable to the recommended clinical exposure (GlaxoSmithKline Pharmaceuticals 2005). In a prior study with sunitinib, rats given 15 mg/kg/day for fourteen days demonstrated significant decreases in body weight (Pfizer 2006; Steeghs et al. 2008). For sorafenib, rats given 25 mg.kg/day for four weeks demonstrated morbidity, whereas a dose of 5 mg/kg/day was well tolerated (Bayer Pharmaceuticals 2005). Based on these findings, male rats (n = 8/group) received twenty-one daily oral doses of vehicle, pazopanib (300 mg/kg/day), sunitinib (10 mg/kg/day), or sorafenib (10 mg/kg/day). Cardiac structure and function were determined using echocardiography before and during a pharmacologic stress test using a continuous infusion of dobutamine prior to the initiation of dosing and on days 14 and 21 of dosing. A separate group of rats (n = 3/group) received seven daily oral doses of pazopanib, sunitinib, or sorafenib to assess the toxicokinetics of each compound and to quantify plasma cardiac troponin levels.
Echocardiography Measurements
Animals were lightly anesthetized using isoflurane delivered in 100% oxygen for both induction (1.5–3%) and maintenance (1-2%). A twenty-four–gauge catheter was placed into a lateral tail vein for the dobutamine or saline (baseline image) infusion. For baseline cardiac structure and function determinations, short-axis images of the LV at the mid-papillary level, and LV long-axis images were obtained and stored as digital video loops. End diastolic volume (EDV) and end systolic volume (ESV) were calculated using the area/length formula (Schiller et al. 1989). Stroke volume (SV), cardiac output (CO), and percentage ejection fraction (EF) were calculated. Fractional area change (FAC) was calculated from the end diastolic and end systolic left ventricular cross-sectional areas and expressed as a percentage change in area. Peak systolic circumferential (SC) and radial strain (SR) and circumferential (SrC) and radial (SrR) strain rates were obtained using two-dimensional gray-scale speckle tracking. Pulsed-wave Doppler was obtained from transmitral and aortic outflow region to determine peak early diastolic E wave velocity and heart rate (Weyman 1994). Left ventricular septal myocardial tissue Doppler was obtained to determine peak diastolic myocardial septal annulus velocity (E′). Images were acquired and stored using the GE Vivid 7 ultrasound system (GE Medical Systems, Milwaukee, WI). Images were analyzed offline using the GE Echo-Pac system (GE Medical Systems).
Two-dimensional and Doppler echocardiographic measurements, as well as calculated parameters, are provided in Figure 1 and as supplementary data (supplementary data is available at http://tpx.sagepub.com/supplemental). For all calculated echocardiographic parameters, group means with the standard error of the mean (SEM) were calculated using SAS for Windows, release 8.2 (SAS Institute, Cary, NC, 1990).
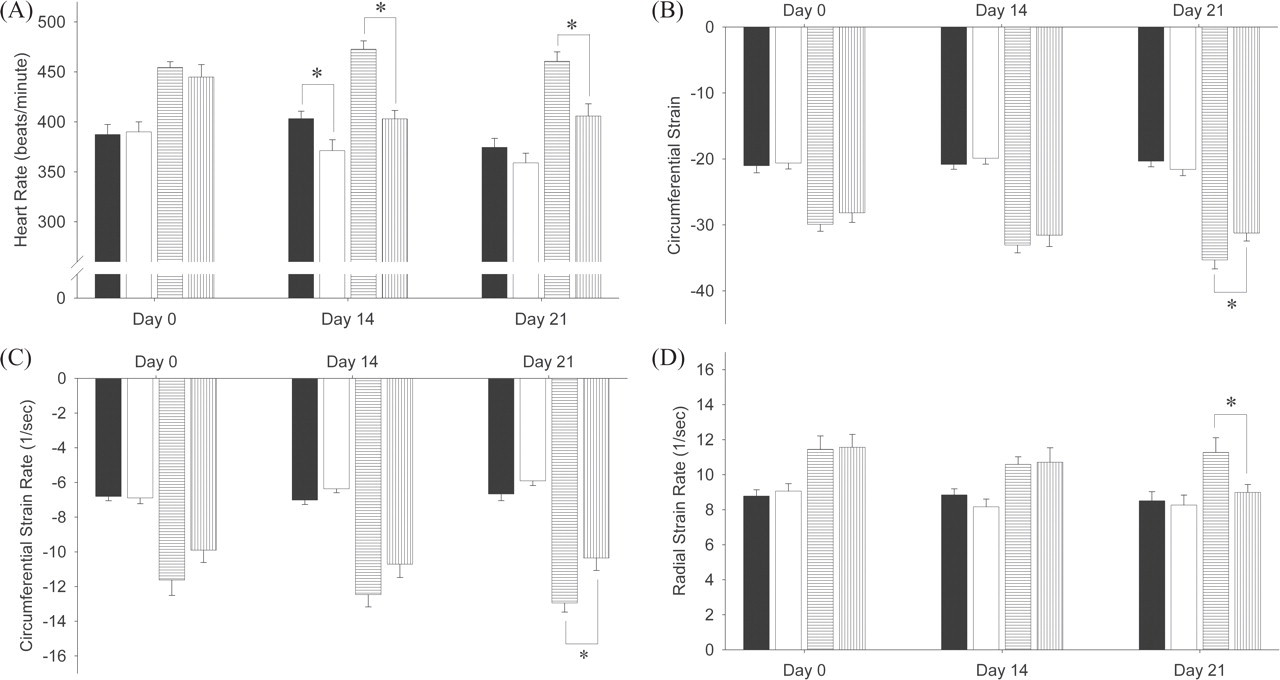
Effect of sorafenib on heart rate (A), circumferential strain (B), circumferential strain rate (C), and radial strain rate (D). Vehicle (filled and horizontal hashed bars) or sorafenib (blank and vertical hashed bars) was administered orally by gavage to conscious rats for twenty-one consecutive days. Echocardiogrpahic parameters were measured before (filled and blank bars) or during (hashed bars) dobutamine infusion. Each value is the mean of eight rats, except for day 21 (n=7 for the sorafenib-treated group). *, Mean value statistically different from the mean value of vehicle group (p < .05).
Terminal Necropsies and Microscopic Examination
Except for one animal given sorafenib that was found dead on day 20 of dosing and the toxicokinetic group, all animals were euthanized via exsanguination under isoflurane anesthesia on day 22 following completion of echocardiography measurements. There were no differences in body weight in treated groups when compared to their respective vehicle-treated cohorts. The hearts were removed, weighed, and bisected longitudinally. One-half was collected and fixed for histopathological evaluation, and the remaining half was collected and fixed for transmission electron microscopy. Samples designated for light microscopic examination were fixed for eighteen to twenty-four hours, then transferred to 70% ethanol, trimmed, processed, and embedded in paraffin wax. Slides were prepared and stained with hematoxylin and eosin (H&E).
Immunohistochemistry for Cardiac Troponin I and TUNEL
Formalin fixed paraffin embedded sections were placed on the Ventana Discovery XT (Ventana Medical Systems, Tucson, AZ). Sections were deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval using an ethylenediaminetetraacetic acid (EDTA)-based antigen retrieval buffer (CC1, Ventana Medical Systems). Sections were blocked with 3% hydrogen peroxide and protein block (Dakocytomation, Carpenteria, CA) with 5% normal goat serum. A mouse monoclonal anti-human troponin I antibody (Chemicon Intl, Temecula, CA) was applied at a concentration of 1 μg/mL for one hour. Omitting primary antibody and using an isotype-matched mouse IgG (Southern Biotechnology) provided the negative control. Primary antibody was labeled using mouse Ultra MAP-HRP polymer (Ventana Medical Systems). Slides were then stained with 3,3-diaminobenzidine (DAB), counterstained with hematoxylin, and coverslipped. With the exceptions noted below, TUNEL assays were performed using the ApopTag peroxidase In Situ Apoptosis Kit (Chemicon Intl, Temecula, CA). Briefly, sections were deparaffinized, rehydrated, and incubated in protease 1 and then blocked with 3% hydrogen peroxide, followed by incubation with equilibration buffer. Sections were then incubated with terminal deoxynucleotidyl transferase (TdT) enzyme and labeled nucleotide substrate to catalyze incorporation of digoxigenin-conjugated nucleotides to DNA fragments. Finally, sections were treated with an anti-digoxigenin biotinylated secondary (Innogenex, San Ramon, CA); streptavidin-HRP; 3,3-diaminobenzidine; and counterstained with hematoxylin.
Cardiac Ultrastructure Using Transmission Electron Microscopy
One-half from each rat heart obtained at the terminal necropsy was fixed (2.5% glutaraldehyde, 2% formaldehyde in 0.1M cacodylate buffer [buffer], pH 7.3) for one hour. Unless noted otherwise, all processing was performed at room temperature.
Samples were collected from atria and ventricles, trimmed to approximately 1 mm3 pieces, and fixed overnight with rocking. Samples were rinsed with buffer for three hours (three 1-hour changes), post-fixed with 1% osmium tetroxide in buffer for two hours, rinsed with buffer for thirty minutes (three 10-minute changes), and dehydrated with a graded series of ethanols (30–100%, thirty minutes each). Samples were infiltrated with a 1:1 mixture of 100% ethanol and epoxy resin for four hours, followed by three 2-hour and one 8-hour infiltration steps in 100% resin. Samples were placed into uniquely labeled molds with 100% resin and polymerized in a 65°C oven overnight. Sections of tissue approximately 0.5 μm thick were cut, collected on glass slides, and stained with 1% aqueous toluidine blue. One ventricle section per rat was examined from all test article–treated rats and from three rats for each concurrent vehicle control group. Regions of interest were identified by light microscopic examination of the stained sections. Subsequently, thin sections of the regions of interest (approximately 90 nm) were cut and collected on copper grids, stained with uranyl acetate and lead citrate, and examined with an FEI Tecnai 20 transmission electron microscope operating at 120 kV. At least twenty-five myofibers from each section were examined. Representative digital images were captured using a Gatan UltraScan 1000 camera.
Evaluation of Plasma Cardiac Troponin I
Approximately 50 μL of plasma obtained from toxicokinetic animals and from an additional six naïve rats were stored frozen at -20°C or lower until time of measurement. Cardiac troponin I (cTnI) was measured in plasma using a commercial rat-specific immunoassay (Meso Scale Discovery [MSD], Gaithersburg, MD) according to manufacturer specifications.
Toxicokinetics of Pazopanib, Sorafenib, Sunitinib, and N-Desethyl Sunitinib
Blood samples (approximately 0.3 mL) were drawn from an appropriate vessel of each toxicokinetic animal. Samples were collected at approximately one, two, four, eight, and twenty-four hours after dosing on day 7. All samples were collected into tubes containing heparin (sorafenib or sunitinib) or EDTA (pazopanib), placed on water ice immediately after collection, and centrifuged. Fifty microliters of the resultant plasma was transferred to a ninety-six–well autosampler tube, frozen, and stored at -20°C or lower prior to analysis. Samples were analyzed using analytical methods based on protein precipitation, followed by HPLC/MS/MS analysis. The lower limit of quantification for GW786034 was 100 ng/mL using a 20 μL aliquot of rat plasma with a higher limit of quantification (HLQ) of 50,000 ng/mL. For sunitinib and N-desethyl sunitinib, the LLQs and HLQs were 10 and 10,000 ng/mL, respectively; for sorafenib, the LLQ and HLQ were 50 and 25,000 ng/mL, respectively (50-μL sample).
Analysis of Data
Group means and the SEM are presented for all quantitative data. All statistical analyses were performed using an end point α level of 5% against the two-sided alternative (i.e., treatment not equal to concurrent vehicle control). A t test (Gosset 1908) using PROC TTEST in SAS was performed on each parameter at each time point, as indicated. If the homogeneity of the variances assumption was satisfied, a parametric analysis was performed; otherwise, a t test with Satterthwaite’s approximation of the degrees of freedom was performed (Heiberger and Holland 2004).
All statistical analyses were performed using SAS for Windows, release 9.1 (SAS Institute, Cary, NC, 2004).
Toxicokinetic analysis was performed by noncompartmental pharmacokinetic analysis using WinNonlin, Enterprise Edition, version 4.1 (Pharsight, St. Louis, MO, 2003). The systemic exposure was determined by calculating the area under the plasma concentration time curve (AUC) from the start of dosing to the last quantifiable time point (AUC0-t) using the linear logarithmic trapezoidal rule. The maximum observed peak plasma concentration (Cmax) and the time at which it was observed (Tmax) were determined by WinNonLin.
In Vitro Cardiomyocyte Assays
Primary cultured neonatal (day 1) rat cardiomyocytes were purchased from Cell Applications, Inc. (San Diego, CA). Cardiomyocyte cultures were provided in serum-free culture medium by the manufacturer preplated in fetal bovine serum (FBS)–coated Whatman Clear View plastic-bottom, ninety-six–well plates at a density of 1 × 104 cells/well and were examined and dosed upon receipt. Cells were exposed to test articles (pazopanib, sunitinib, or sorafenib at concentrations of 1 and 4 μM) or positive controls (FCCP and doxorubicin) in serum-free medium for forty-eight hours. Viability, apoptosis, and mitochondrial membrane potential (MMP) were assessed using Hoechst 33342 (HO, cell-permeable DNA-binding dye), TO-PRO-3 (Annexin V-fluorescein isothiocyanate [FITC]), and tetramethylrhodamine methyl ester (TMRM), respectively, which were purchased from Invitrogen-Molecular Probes (Eugene, OR). End points were measured via fluorescence multiplexing on an iCyte Imaging Laser Scanning Cytometer (CompuCyte, Cambridge, MA). Hoechst 33342 was excited at 405 nm and emission fluorescence detected via a 463/39 nm bandpass filter. Annexin V-FITC and TMRM were excited at 488 nm and emission fluorescence detected via 530/30 nm and 580/30 nm bandpass filters, respectively; TO-PRO-3 was excited at 633 nm and emission collected via a 650 nm longpass filter. Intracellular ATP levels were measured using the ATP Bioluminescent Somatic Cell Assay Kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Luminescence was monitored using a Gemini XS SPECTRAmax dual-scanning microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA) in luminescence mode. Cell viability was determined by calculating the percentage of cells staining positive for TO-PRO-3 staining. Apoptotic cells were defined as TO-PRO-3 negative/Annexin V-FITC positive. Effects on MMP in cardiomyocytes were measured by mean fluorescence intensity change of TMRM in treated samples compared with corresponding vehicle control.
Results
Toxicokinetics of Pazopanib, Sorafenib, Sunitinib, and N-Desethyl Sunitinib
Doses were chosen based on previously published data in the public domain (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm) and, based on tolerability data from subacute studies in rats and a bridging single-dose toxicokinetic study, were standardized to be as close to the maximum tolerated dose as possible for a twenty-one–day study. One rat given sorafenib was found dead on day 20, whereas all others survived until study termination.
Following seven days of repeat oral administration of pazopanib, sorafenib, or sunitinib, all test articles, including the metabolite N-desethyl sunitinib, were quantifiable in the plasma of all animals for the entire twenty-four–hour sampling occasion (Table 1). Maximum plasma concentrations were observed between two and eight hours after dosing. The systemic exposures achieved in this study following seven daily doses of each test article were comparable to those achieved following single or subacute oral dosing regimens in prior toxicity studies in rats (Bayer Pharmaceuticals 2005; Pfizer 2006). Additionally, Pazopanib exposure levels were at least five-fold above that achieved with sunitinib or sorafenib.
Toxicokinetic summary of repeat dosing of pazopanib, sorafenib, and sunitinib in rats.
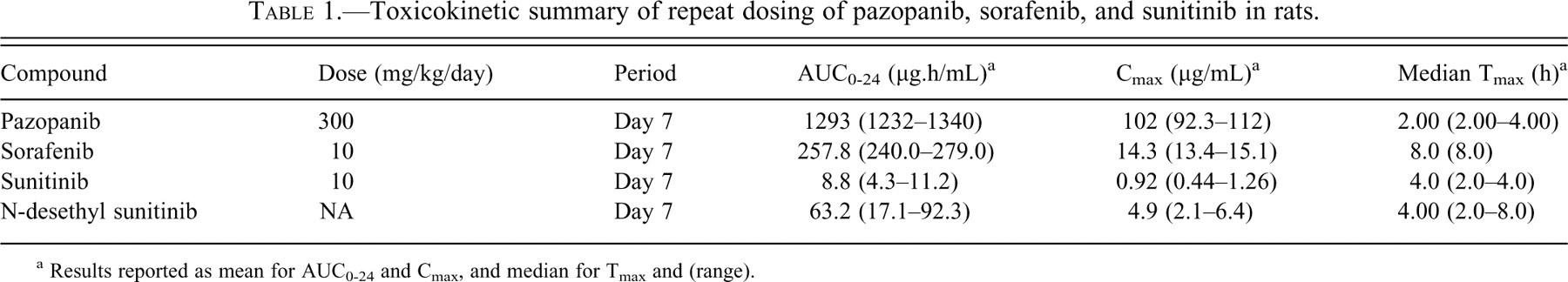
a Results reported as mean for AUC0-24 and Cmax, and median for Tmax and (range).
Echocardiography
Following fourteen or twenty-one daily doses of sunitinib, there were no significant differences in baseline heart rate (HR), EDV, ESV, SV, CO, SR, SC, SrR, and SrC from the concurrent vehicle control group (data not shown). There were no differences in the baseline diastolic parameters, E, isovolumic relaxation time (IVRT), E′, and E-to-E′ ratio between the vehicle and treated groups. A statistically significant increase in ejection fraction (EF) was evident on days 14 and 21 following sunitinib treatment as compared to the concurrent vehicle control group. However, because there were no differences in EDV, SV, and FAC as compared to the concurrent vehicle control, the change in EF was not considered to be related to treatment. Dobutamine infusion increased HR, CO, SV, EF, SC, SrC, SR, and SrR and decreased ESV in both the vehicle and sunitinib-treated groups. Although there was a statistically significant increase in dobutamine-induced circumferential strain on day 14 in the treated group versus the concurrent vehicle control, this difference was not evident on day 21 and was not supported by any changes in additional functional parameters such as EF, CO, and FAC on this day.
Following fourteen daily doses of sorafenib, there was a significant decrease in HR in the treated group (Figure 1A). There were no apparent differences in the EDV, ESV, SV, CO, EF, SR, SrR, SC, SrC and baseline diastolic parameters, E, IVRT, E′, and E-to-E′ ratio between the vehicle and sorafenib-treated groups (data not shown). Dobutamine infusion increased HR, CO, SV, EF, SC SrC, SR, and SrR and decreased ESV in both the vehicle and sorafenib-treated groups. At day 21, there was a statistically significant increase in baseline FAC in the sorafenib group. However, because there were no differences in EDV, SV, and EF as compared to the concurrent vehicle control, this difference is not considered to be related to test article treatment. At day 14, the dobutamine-induced increase in HR was significantly less in the sorafenib-treated group (Figure 1A), and at day 21, the dobutamine-induced increases in HR, SC, SrC, and SrR were significantly decreased compared to vehicle group (Figures 1A, 1B, 1C, and 1D, respectively). In addition, one rat was noted as having multiple premature ventricular complexes during the echocardiography measurements on day 21 (data not shown).
Following fourteen or twenty-one daily doses of 300 mg/kg/day pazopanib, there were no significant differences in any functional parameters from the concurrent vehicle control group.
There were no differences in the baseline diastolic parameters E, IVRT, E′, and E-to-E′ ratio between the vehicle and the treated groups (data not shown). Dobutamine infusion produced the expected increases in HR, CO, SV, EF, SC, and SrC, SR, and SrR and decrease in ESV in both the vehicle and pazopanib-treated groups.
Cardiac Ultrastructure
By transmission electron microscopy, ultrastructural changes in ventricular myofibers in rats after twenty-one days of dosing were limited to degenerative changes in mitochondria in rats given sunitinib or sorafenib. Mitochondrial changes were observed in six of eight rats given sunitinib and included disruption and/or loss of cristae, cavitation of matrix, swelling, and increased numbers of electron-dense matrix deposits (Figure 2).
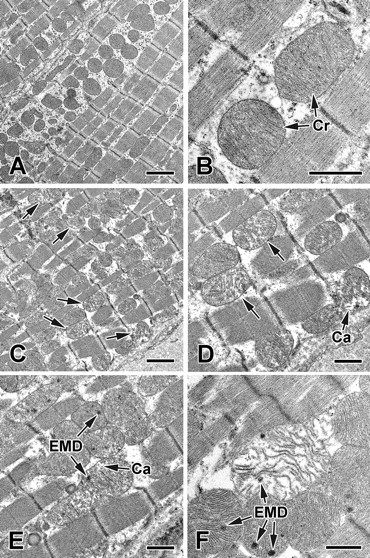
Representative transmission electron micrographs of ventricular myofibers from rats given sunitinib. (A and B) Micrographs of ventricular myofibers from rat given vehicle; (C–F), images from rat given 10 mg/kg/day sunitinib for twenty-one days. Note numerous cristae in mitochondria of the control rat (Cr, in B) and loss and/or disruption of cristae in mitochondria of affected rats given sunitinib (arrows, in C and D). Mitochondrial degenerative changes including matrical cavitations (Ca, in D and E), numerous electron-dense matrical deposits (EMD, in E and F), and swelling (in F) were seen in rats given sunitinib. Bars = 2 μm (A and C). Bars = 1 μm (B, D, E, F).
In rats given sorafenib, mitochondrial degeneration was observed in only one of seven animals. Degeneration was characterized by disruption and/or loss of cristae and swelling, whereas cavitation of matrix and electron-dense matrix deposits were rarely observed (Figure 3).
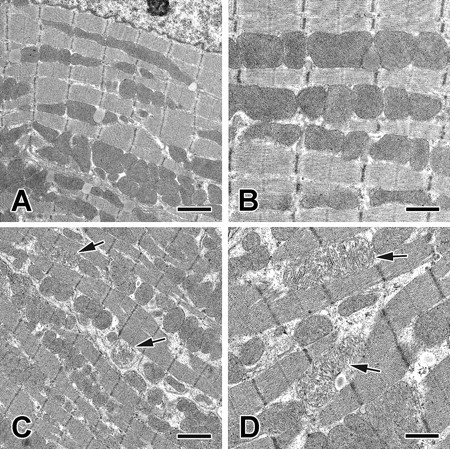
Representative transmission electron micrographs of ventricular myofibers from rats given sorafenib. (A and B) Micrographs of ventricular myofibers from rat given vehicle; (C and D) images from rat given 10 mg/kg/day sorafenib for twenty-one days. Note loss and/or disruption of mitochondrial cristae of affected rat given sorafenib (arrows, in D). Bars = 2 μm (A and C). Bars = 1 μm (B and D).
No ultrastructural changes were seen in mitochondria or in myocardiocytes from rats given pazopanib (data not shown).
Histopathology and Plasma cTn I
There were no test article–related macroscopic or light microscopic findings noted in the hearts of any animals given sunitinib, sorafenib, or pazopanib. Microscopic findings consistent with the spontaneous background lesion of cardiomyopathy were present in the hearts of multiple rats across dose groups, including those given vehicle. These lesions were characterized by small aggregates of mononuclear inflammatory cells associated with focal areas of myocardial degeneration. These areas were also associated with focally increased numbers of TUNEL–positive cells by immunohistochemistry, and generally with loss of troponin I immunostaining. Cardiomyopathy is common in rats of this age and sex, occurred in approximately equal incidence across groups and, therefore, was not considered related to administration of any of the angiogenic compounds. With the exception of the foci of cardiomyopathy, there were no significant qualitative or quantitative differences in either TUNEL immunostaining or troponin I immunostaining in the cardiomyocytes of rats given any of the vehicles or sunitinib, sorafenib, or pazopanib. TUNEL staining was generally very minimal and widely scattered across the heart in individual cells. Similarly, troponin I immunostaining was absent within foci of cardiomyopathy, but otherwise was similar in all rat hearts examined.
Abnormalities in plasma cTn I collected approximately twenty-four hours after dosing on day 7 were not noted in animals given any of the test article compounds in this study, with all values equal to or below levels found in naïve controls, which ranged from 0.020 to 0.092 μg/L (data not shown).
Heart Weights
There was a statistically significant decrease in mean heart weight (p < .05), normalized to body weight in both the sunitinib- and pazopanib-treated animals as compared to their concurrent vehicle control groups. These differences, however, were minor, and the individual heart weights for the sunitinib- and pazopanib-treated groups were within the range of heart weights observed in the three vehicle control groups. In addition, left ventricle mass for the sunitinib- and pazopanib-treated groups did not correlate with the changes observed in the heart weights for these groups. Therefore, the differences in normalized heart weights were not considered to be drug related and instead were most likely a result of animal variability within the treatment groups.
Cardiomyocyte Culture Assay
To gain a better understanding of the in vivo findings, we evaluated the effects of pazopanib, sunitinib, and sorafenib on mitochondrial membrane potential (MMP), viability, apoptosis, and ATP levels in cultured neonatal rat cardiomyocytes using laser scanning cytometry. None of compounds demonstrated changes in viability at tested concentrations, whereas positive controls produced robust cytotoxicity. No significant amount of apoptotic cells (TO-PRO-3 negative/Annexin V-FITC positive) were identified in any samples. Similarly, no changes were detected in any parameters following treatment for twenty-four hours (data not shown).
The effects on MMP of cardiomyocytes after forty-eight hours of exposure were quantified and are illustrated in Figure 4. Pazopanib and sorafenib had no effect on MMP, whereas sunitinib induced a slight but significant decrease in MMP in a dose-dependent manner. As a positive control, FCCP dose-dependently depolarized mitochondria. Doxorubicin also significantly decreased MMP in both populations. Effects on intracellular ATP levels are summarized in Figure 5. Pazopanib did not significantly change ATP levels, whereas both sunitinib and sorafenib induced slight but significant decreases in ATP levels (as low as 80% of control) in a dose-dependent manner. As expected, FCCP and doxorubicin robustly depleted intracellular ATP.
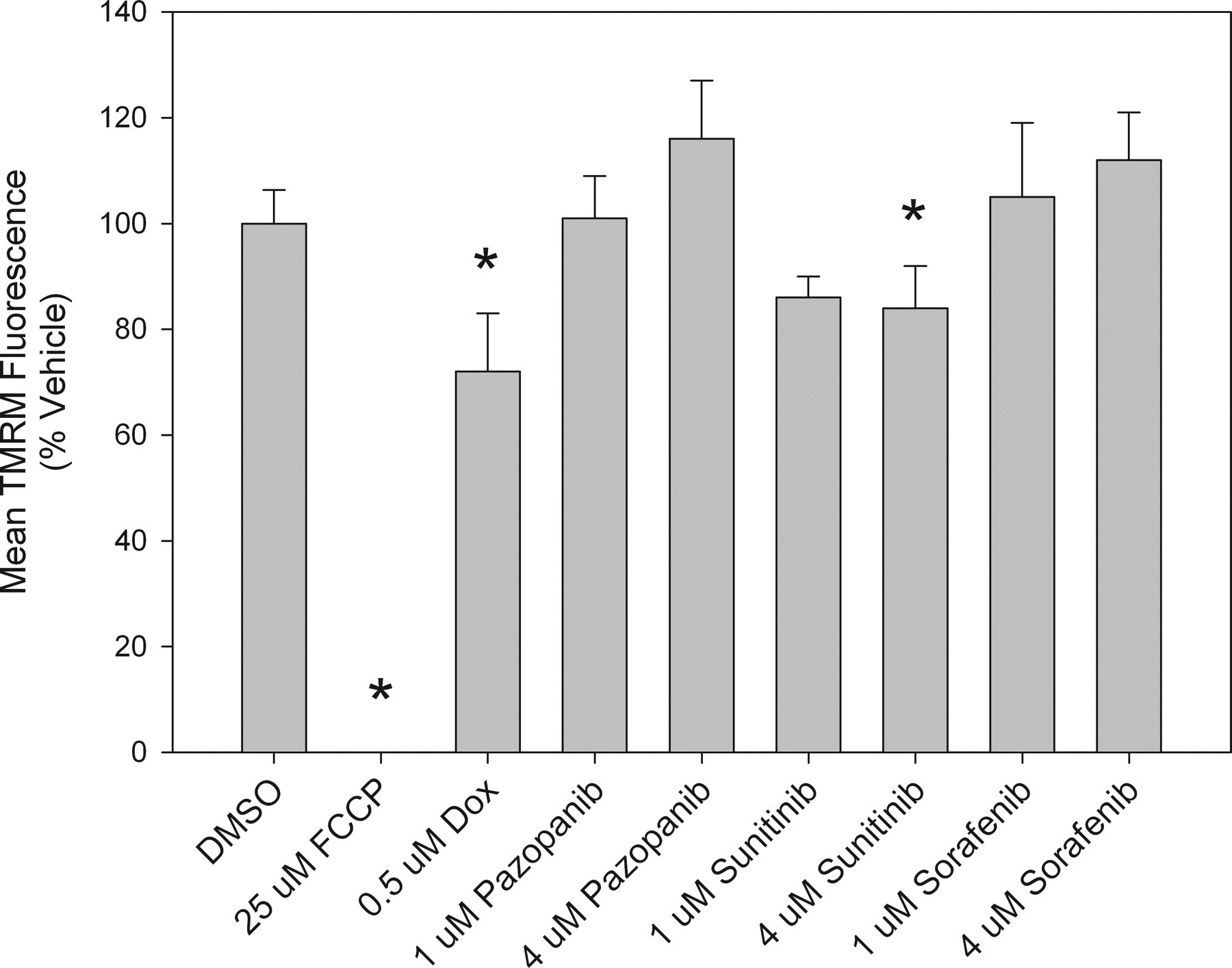
Mitochondrial membrane polarization changes in rat cardiomyocytes. Data represent values in cultures following forty-eight–hour exposure. All values represent the mean ± standard deviation, n = 4. Asterisks indicate values significantly different from the correspondent vehicle control (p < .05).
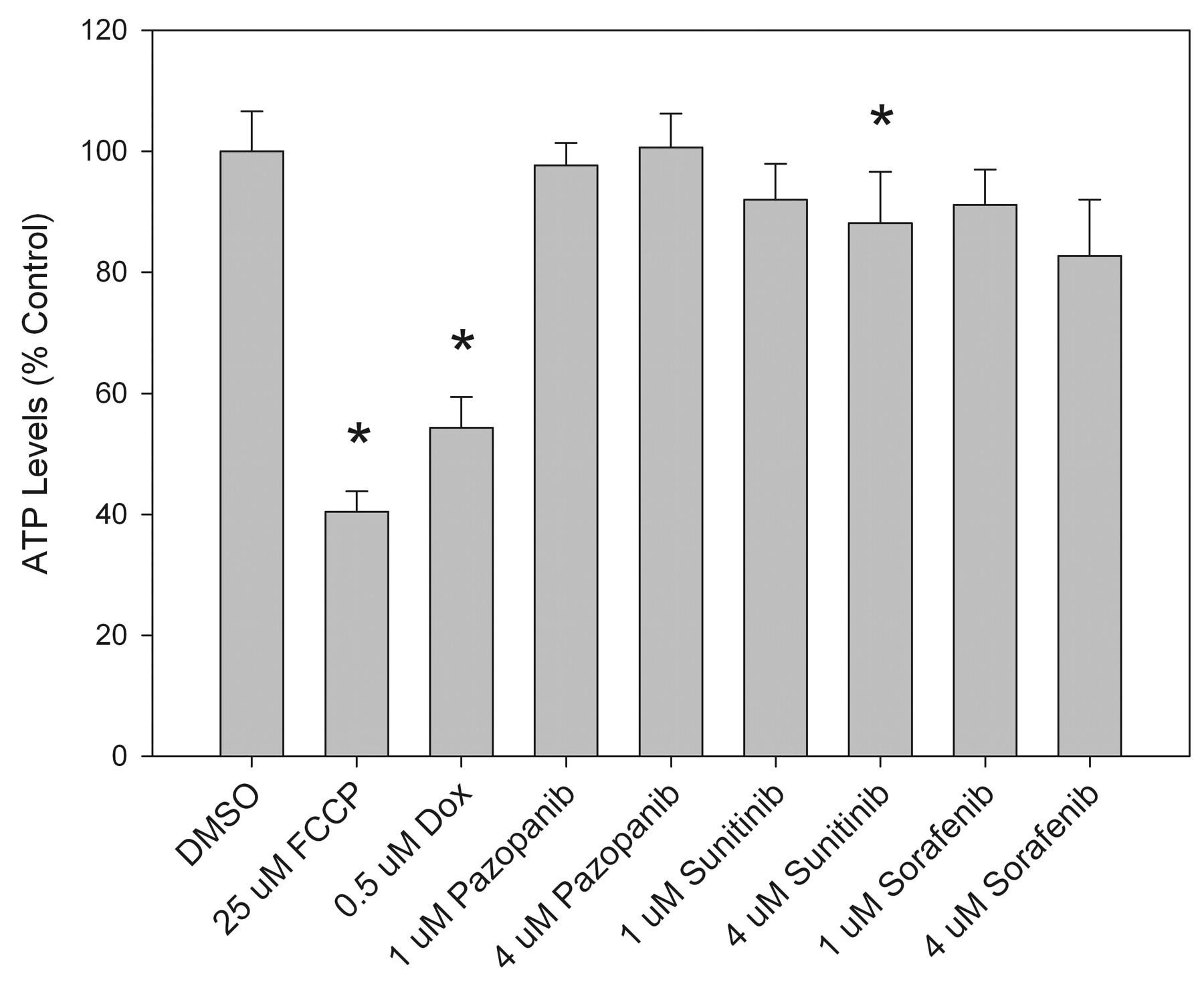
Intracellular ATP levels in rat cardiomyocytes. Data represent values in cultures following forty-eight–hour exposure. All values represent the mean ± standard deviation, n = 8. Asterisks indicate values significantly different from the correspondent vehicle control (p < .05).
Discussion
In this study, we evaluated the effects of a panel of multikinase angiogenic inhibitors on myocardial function and structure. In previously published studies, VEGFr inhibitors have demonstrated mitochondrial and/or cardiovascular toxicity, suggesting the possibility of an on-target pharmacologic effect of this class of anti-angiogenic agents. Although minimal heart lesions have been noted with sunitinib (myocardial vacuolation, pericardial inflammatory infiltrates) and sorafenib (myocardial inflammatory infiltrates) in chronic preclinical rat toxicologic studies (none has been noted with pazopanib), it is important to note that no histologic lesions should be expected in the heart in subacute studies with any of these compounds. In this study, we dosed rats with sunitinib, sorafenib, or pazopanib for twenty-one days and assessed cardiac functional effects using dobutamine stress echocardiography coupled with structural myocardial analyses. We chose maximally tolerated doses for each compound and achieved exposures at or above levels achieved in clinical studies (Pfizer 2007; Strumberg et al. 2007). Additionally, pazopanib exposures significantly exceeded those attained with sunitinib or sorafenib treatment. Overall, neither pazopanib nor sunitinib had apparent effects on baseline systolic and diastolic cardiac function or dobutamine-induced increases in cardiac function. Interestingly, with sorafenib treatment, we observed premature ventricular contractions, as well as alterations in HR, SR, SrR, and SrC following dobutamine challenge on day 21 of dosing. This finding suggests that sorafenib can decrease both conduction and left ventricular contractility. Echocardiography changes, including changes in contractility, rhythm, conduction, axis, QRS amplitude, ST changes, T wave changes, and QT prolongation, have been reported in patients given sorafenib (Schmidinger et al. 2008). Additionally, each compound has been evaluated for effects on ion channels and action potential in prior work. Sunitinib demonstrated binding and/or inhibitory potential at clinically relevant levels toward serotonin receptor 5HT2a, a 1 b -adrenergic receptor, hERG, and Purkinje fiber action potential (Pfizer 2006), whereas sorafenib inhibited hERG-dependent current and Purkinje fiber action potential at concentrations exceeding clinical exposures and exposures attained in this study (Bayer Pharmaceuticals 2005). Pazopanib, however, demonstrated minimal inhibition of hERG channel and did not inhibit Purkinje fiber action potential. Thus, the electrocardiogram effects seen with sorafenib in this study cannot be readily attributed to ion channel effects.
The dobutamine stress assay is a clinically important and relevant tool for assessing myocardial function. This approach has been successfully used to detect changes in cardiac function in long-term survivors of anthracycline chemotherapy (Klewer et al. 1992; Paiva et al. 2005), and our findings with sorafenib indicate the potential for impairment of cardiac reserve with treatment. The rat is considered to be an appropriate model for assessing cardiomyopathies, as it has been shown to recapitulate doxorubicin-induced cardiomyopathy observed in patients (Burdick et al. submitted for publication). Because simply giving sorafenib, sunitinib, or pazopanib for twenty-one days to rats did not result in any histologic evidence of myocardial toxicity, the use of a cardiac stressor such as dobutamine infusion is necessary to elucidate drug-induced functional effects. A separate strategy for inducing cardiac stress in rodents has been previously performed with sunitinib using phenylephrine (Chu et al. 2007).
The ultrastructural changes observed in rats given sunitinib or sorafenib indicate that cardiac mitochondria may be a target of toxicity. Myocardial mitochondrial degeneration has been observed with multiple chemotherapeutic agents (Kerkela et al. 2006; Wallace 2003), and in particular, direct effects of sorafenib on rat heart mitochondria have been previously characterized (Will et al. 2008). The changes we noted with sorafenib occurred in parallel with functional observations, but the structural mitochondrial changes associated with sunitinib occurred in the absence of functional changes. It is likely that the difference in ultrastructural observations between the two, along with the different responses to dobutamine stress, are a function of disparate mechanisms of toxicity based on differences noted in the in vitro assays. Mitochondria play a key role in cardiac reserve (Hanley et al. 2002; McCarthy et al. 2005), and drug-induced mitochondrial dysfunction may impair the response to a stressor such as dobutamine.
Angiogenic inhibitor effects on neonatal rat cardiomyocyte cultures were assessed using multiparameter laser scanning cytometry. Neonatal rat cardiomyocytes have been shown by others to express key angiogenic genes, including VEGFR (Zentilin et al. 2009), PDGFR (Yano et al. 2008), and c-kit (Tallini et al. 2009), and as such have been used as an in vivo surrogate to assess effects on angiogenic signaling modulation in cardiomyocytes. Although all three compounds are potent inhibitors of angiogenic signaling pathways, different effects were observed between compounds. Pazopanib did not affect any changes on ATP, MMP, or cell viability up to its limit of solubility. Sunitinib caused mitochondrial depolarization and ATP depletion without affecting viability in cardiomyocytes. When compared to the ultrastructural mitochondrial effects observed in vivo, these changes were milder than expected, even at comparatively higher concentrations. Although the reason is unknown, the difference may simply be owing to the much longer exposure period (twenty-one days) in vivo as compared to the in vitro experiment (forty-eight hours). TMRM has been shown to be one of the more sensitive markers for mitochondria in neonatal rat cardiomyocytes (Hattori et al. 2010), and therefore the discrepancy is not likely a result of marker selection. These results are in concordance with prior findings by Chu et al. (2007), who demonstrated that comparable concentrations of sunitinib induced mitochondrial cytochrome c release and apoptosis in cardiomyocyte cultures, and the findings of Will et al. (2008), who demonstrated ATP depletion in the embryonic rat heart cell line H9c2. None of the compounds induced changes in MMP, viability, or apoptosis in noncardiomyocytes in culture (data not shown). The differences in response between cardiomyocytes and noncardiomyocytes, as hypothesized by others (Kerkela et al. 2006), are most likely owing to altered kinase expression profiles or different requirements for oxidative phosphorylation and ATP for cardiomyocytes. When combined, the findings from this study and from other groups demonstrate agent-specific cytotoxic effects on myocardial cell culture, initially characterized by ATP depletion and followed by events associated with activation of the apoptotic pathway (mitochondrial membrane depolarization, cytochrome c release). Both sunitinib and sorafenib decreased ATP levels, which has also been observed by others at comparable exposures (Will et al. 2008). In the same study, sorafenib was shown to be a potent mitochondrial uncoupler and, at higher concentrations, a Complex V inhibitor. These direct effects on mitochondrial function may have contributed to sorafenib-induced ATP depletion observed in this study. The exact mechanism of sorafenib-induced ATP depletion in cardiomyocytes is unknown, but it could be related to inhibitory effects on other kinases or inhibition of mitochondrial function.
Given the varied outcomes of the functional and structural cardiac studies with each compound, these findings suggest that the potential for myocardial toxicity is not directly related to the anti-VEGFr2 pharmacologic activity of these agents and that the underlying mechanism of myocardial injury may differ among agents. Several lines of evidence highlight potential antiangiogenic-independent mechanisms of sunitinib and sorafenib. Cardiotoxicity mediated through alternative kinase inhibitory pathways has already been implicated for sunitinib and sorafenib via both RSK and AMPK and both RAF1 and BRAF, respectively (Force et al. 2007; Orphanos et al. 2009). Although all three compounds share similar potencies to VEGFr-2, platelet-derived growth factor receptor-β, and c-kit, several groups have identified significant differences in off-target kinase inhibition profiles (Karaman et al. 2008; Kumar et al. 2009). Kumar demonstrated that sunitinib had the broadest kinase activity, with 20% of the kinases inhibited by > 50% at 0.3 μM, whereas pazopanib and sorafenib inhibited 12% and 11% of the tested kinases, respectively. Although there was some overlap identified in the specific kinases inhibited by pazopanib and sorafenib, there are also clear distinctions for some of the kinases. For instance, although pazopanib and sorafenib both inhibit BRAF and CRAF (RAF1) kinases, sorafenib has more than twenty-five–fold higher affinity for RAF kinases than pazopanib, and this target has been mechanistically linked with cardiotoxicity (Yamaguchi et al. 2004). There are likely several other key kinases whose inhibition may lead to myocardial dysfunction.
This is not to say that all toxicities from antiangiogenic therapies are owing to off-target effects. Both biologic and small molecule therapies targeting VEGF and VEGFr have demonstrated clinical side effects including hypertension, gastrointestinal toxicity, proteinuria, and coagulation disorders (Roodhart et al. 2008; Sica 2006). Hypertension, one of the most common adverse clinical effects noted with the antiangiogenic TKI therapies, has been mechanistically linked with VEGFr inhibition and is hypothesized to involve nitric oxide (Ku et al. 1993), vascular rarefaction, or increased arterial stiffness (Veronese et al. 2006). However, it has been difficult to evaluate in preclinical species because of embryonic lethality in knockout models and was not noted in preclinical toxicologic studies (Carmeliet et al. 1996; Ferrara et al. 1996; Patyna et al. 2008). Therefore, although hypertension may play a role in some of the cardiac and cardiovascular adverse events noted with these agents in human clinical trials, it does not appear to be associated with myocardial and mitochondrial changes that are described in the present set of studies. Rather, assessing functional parameters in cardiac stress models, such as the dobutamine infusion performed in this study, can detect drug-induced changes with higher sensitivity than standard techniques. Differences in the potential for myocardial injury among TKI’s are not only apparent with in vitro and rodent studies, but also from data from clinical trials. Cardiac adverse events have not been noted equally among agents and have been well documented for sunitinib and sorafenib. In a Phase III trial, 10% of patients given sunitinib had decreased LV EF (Motzer et al. 2007), and a retrospective analysis of 224 patients who received sunitinib over a one-year period identified a 6% rate incidence of symptomatic heart failure, with a strong correlation with a history of hypertension (Khakoo et al. 2008). For sorafenib, 3% of patients in a Phase III trial developed cardiac ischemia (Escudier et al. 2007). A prospective study with RCC patients given sunitinib or sorafenib, 34% experienced increased cardiac enzymes, arrhythmia, left ventricular dysfunction, or acute coronary syndrome (Schmidinger et al. 2008). In a Phase III trial with bevacizumab, a low but significant incidence of cardiac ischemia was observed (Rini et al. 2008). The clinical cardiotoxic profile of pazopanib is promising, with reports of cardiac adverse events (other than hypertension) extremely rare in the approximately 2,000 patients examined up to the time of regulatory filing. This finding is consistent with the negative preclinical toxicity data, as well as the rat dobutamine stress data and in vitro mitochondrial data generated in this study. However, the lack of clinical cardiotoxicity cases should be viewed with caution and require continued evaluation owing to pazopanib’s very recent regulatory approval.
In conclusion, we have demonstrated that, although pazopanib, sunitinib, and sorafenib have a similar mechanism of action, they differ in their effects on functional and structural parameters of myocardial toxicity. These differences suggest that cardiotoxicity is not necessarily related to a pharmacologic class-wide effect of VEGFR inhibition, and the rat myocardial structural and functional changes identified in this study may be instead a result of inhibition of other kinase pathways, the mechanism of which may be associated with mitochondrial toxicity.
Footnotes
Acknowledgments
The authors wish to thank Rosanna Mirabile, Janice Kane, Karen Lynch, Loren Kohrs, Jeffrey Burdick, Kai-Fen Wang, and Rakesh Kumar for their technical support and guidance.