
Abstract
Acetaminophen (APAP) is a widely used antipyretic and analgesic agent. However, overdosing and sometimes even a recommended dose can lead to serious and conceivably fatal liver toxicity. Therefore, it is important to clarify understand mechanisms of hepatotoxicity induced by APAP. Gap junctions, formed by connexin, have important roles in maintenance of tissue homeostasis and control of cell growth and differentiation. In the liver, Cx32 is a major gap junction protein whose expression is known to gradually decrease with chronic liver disease progression. In the present study, acute hepatotoxic effects of APAP were found to be reduced in Cx32 dominant negative transgenic rats lacking normal gap junctional intercellular communication in the liver. In littermate wild-type rats, the injured centrilobular hepatocytes were positive for TUNEL staining and featured elevated expre ssion of cleaved caspase-3 and Cx43, which is not expressed in normal hepatocytes. These results suggest that APAP hepatotoxicity involves apoptosis, and induction of Cx43 expression may play an important role in the apoptotic signaling. Moreover, gap junctional functions of Cx32 can play important roles in removing damaged hepatocytes by apoptosis for liver tissue homeostasis.
Introduction
Gap junctional intercellular communication (GJIC) plays important roles in tissue homeostasis (Loewenstein 1981), embryonic development (Guthrie and Gilula 1989), and carcinogenesis (Trosko and Chang 2001; Yamasaki 1990). Connexins are subunits of gap junction channels that allow intercellular exchange of small molecules, such as ions, second messengers, and cellular metabolites, between contacting cells (Bennett et al. 1991; Evans and Martin 2002; Gilula, Reeves, and Steinbach 1972; Loewenstein 1981).
Among more than twenty members of the Connexin family, Cx32 and Cx26 are major gap junction proteins in the liver (Paul 1986). Cx32 protein levels decrease during chemical hepatocarcinogenesis in the rat liver (Krutovskikh et al. 1995; Krutovskikh, Oyamada, and Yamasaki 1991; Miyashita et al. 1991) and with progression of human chronic liver disease from cirrhosis to hepatocellular carcinomas (HCCs) (Nakashima et al. 2004).
We previously established two lines of transgenic rats carrying a dominant negative mutant of Cx32 under albumin promoter control, which have much decreased capacity for GJIC as measured by dye-transfer assay in vivo (Asamoto et al. 2004). These transgenic rats (Cx32ΔTg) were found to be resistant to the hepatotoxicity of D-galactosamine and carbon tetrachloride (Asamoto et al. 2004) but more susceptible to both early and late stages of hepatocarcinogenesis, including lung metastasis (Hokaiwado et al. 2007, 2005). Thus, normal functions of Cx32 may play important roles in liver tissue.
Acetaminophen (APAP) is a widely consumed analgesic contained in several nonprescription pharmaceuticals. It is not normally toxic when used at therapeutic levels (O’Grady 1997; Rumack 2004). However, single or cumulative overdose of APAP can cause severe liver injury with the potential to progress to liver failure (Lee 2004; Prescott 1980). Even at normal therapeutic doses, serious liver damage has been reported (Floren, Thesleff, and Nilsson 1987; Seirafi, Iten, and Hadengue 2007). Actually, APAP is the most frequent cause of drug-induced liver failure in the United States and in Great Britain (Lee 2004). Furthermore, fasting and alcohol drinking may enhance APAP-induced hepatotoxicity (Rumack 2004; Whitcomb and Block 1994), and there may also be an increased risk of acute liver injury with hepatitis C virus (HCV) infection in those consuming APAP (Nguyen, Sam, and Thuluvath 2008). However, the relation between gap junction function and APAP hepatotoxicity is unclear.
The present study was therefore carried out to investigate whether the loss of GJIC impacts on APAP-induced acute liver injury. For this purpose, hepatotoxic effects were examined in Cx32ΔTg rats in comparison with wild-type littermates.
Materials and Methods
Production of Transgenic Rats
The establishment, production, and screening of Cx32ΔTg rats carrying mutated Cx32 gene were as previously described in detail (Asamoto et al. 2004). Animals having high copy numbers (~50 copies) of the transgene were used in this study. Cx32ΔTg male rats were produced by mating heterozygous Cx32ΔTg males with wild-type Sprague-Dawley (SD) females (Japan SLC, Shizuoka, Japan). Rats were maintained in plastic cages on hardwood chips, in an air-conditioned, specific pathogen-free (SPF) animal room at 22 ± 2°C and 50% humidity with 12hr/12hr light-dark cycle. All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Nagoya City University School of Medical Sciences.
Experimental Protocols
Cx32ΔTg rats and littermate wild-type rats at ten weeks of age were given a single i.g. injection of 250, 500, or 1,000 mg/kg APAP (Wako, Osaka, Japan), dissolved in 0.5% methylcellulose (Wako). Each dose was given to seven Cx32ΔTg and seven wild-type rats. As vehicle controls, additional groups of five Cx32ΔTg and five wild-type rats were given 0.5% methylcellulose. All rats were sacrificed twenty-four hours after the treatments. Serum samples were collected from the abdominal aorta for measurement of aspartate aminortansferase (AST) and alanine aminotransferase (ALT), and immediately the livers were excised, weighed, and cut into slices for freezing or fixation in 10% buffered formalin and routine processing for embedding in paraffin for histological evaluation (2–3 μm thickness). Sections were routinely stained with hematoxylin and eosin. Frozen liver samples were used for immunohistochemical staining and western blotting. Serum contents of ALT and AST were analyzed using radioimmunoassays by a commercial laboratory (SRL, Inc., Tokyo, Japan).
Histological Analyses
For quantifying hepatotoxicity, histopathological changes in each liver were classified as mild (+1), moderate (+2), severe (+3), or none (0), taking account of the area of cell death, degeneration (ballooning), and inflammation around the central veins (0; 0%, +1; less than 20%, +2; 20 ~ 70%, 3; more than 70% of hepatic lobules) as shown in Figure 2.
Immunohistochemical Staining of Cx32 and Cx43
Detailed methods for fluorescence immunohistochemistry were as described previously (Asamoto et al. 2004). Frozen sections were cut at 5 μm and fixed in cold acetone and 10% buffered formalin. A polyclonal antibody against Cx32 (INVITROGEN, Carlsbad, CA), which recognizes a deleted part of the transgene, and a monoclonal antibody against Cx43 (INVITROGEN) were used with biotin-conjugated anti-rabbit or anti-mouse IgG and TRITC-labeled or FITC-labeled streptavidin (Vector Lab, Burlingame, CA, USA) to visualize both Cx32 and Cx43 endogenous proteins under fluorescence microscopy (Olympus AX-70, Tokyo, Japan).
Analyses for Apoptosis
Apoptotic cells were detected by terminal deoxy nucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay as well as cleaved caspase-3 immunohistochemistry. TUNEL assay was performed using an In Situ Apoptosis Detection Kit from Takara (Otsu, Japan). For detection of cleaved caspase-3, de-paraffinized slide sections of liver tissues were fixed with 10% formalin and then incubated with 1:100 diluted cleaved caspase-3 (Cell Signaling Technology, Boston, MA, USA) antibody. Antibody binding was visualized by a conventional immunostaining method using an auto-immunostaining apparatus (VENTANA HX SYSTEM, VENTANA Japan, Tokyo, Japan).
Western Blotting
Liver tissues were prepared as described previously (Asamoto et al. 2004). Briefly, 10 μg samples per lane were loaded and separated on 12% acrylamide gels and electroblotted onto nitrocellulose membranes (Hybond-ECL, GE Healthcare UK Ltd., Buckinghamshire, UK). Cx43 and cleaved caspase-3 expression levels were assessed using the same antibodies as applied for immunohistochemical staining. β-Actin expression was evaluated to confirm equal amounts of protein loading with a monoclonal anti-β-actin, AC-74 (SIGMA-Aldrich Corp, St. Louis MO, USA).
Results
The treatments with methylcellulose solution alone did not have any influence on livers in either Cx32ΔTg or littermate wild-type rats. With APAP, livers were enlarged and partly necrotic with active bleeding in wild-type rats, especially groups receiving 1,000 or 500 mg/kg (Figure 1A, D). Relative liver weights were increased in a dose-dependent manner, and AST and ALT were dramatically elevated in both Cx32ΔTg and wild-type rats as detailed in Table 1. However, Cx32ΔTg rats demonstrated lower relative liver weights and lower levels of AST and ALT as compared to wild-type rats, and statistical significance (p = .001) was reached for relative liver weights at 500 mg/kg (Table 1). Histopathological examinations revealed massive cell death in centrilobular regions of the livers in almost all wild-type rats (13/14 rats) given 1,000 and 500 mg/kg. With 250 mg/kg, mild centrilobular cell damage with active inflammation and ballooning was observed (Figure 1E, F, and Table 2). These histological changes were graded as described in Materials and Methods (representative samples are shown in Figure 2). In contrast to wild-type rats, more than half of the Cx32ΔTg rats (9/17) did not exhibit any changes in the livers, and the remaining eight rats showed mild to moderate centrilobular cell damage with inflammation (Figure 1B, C, Table 2).
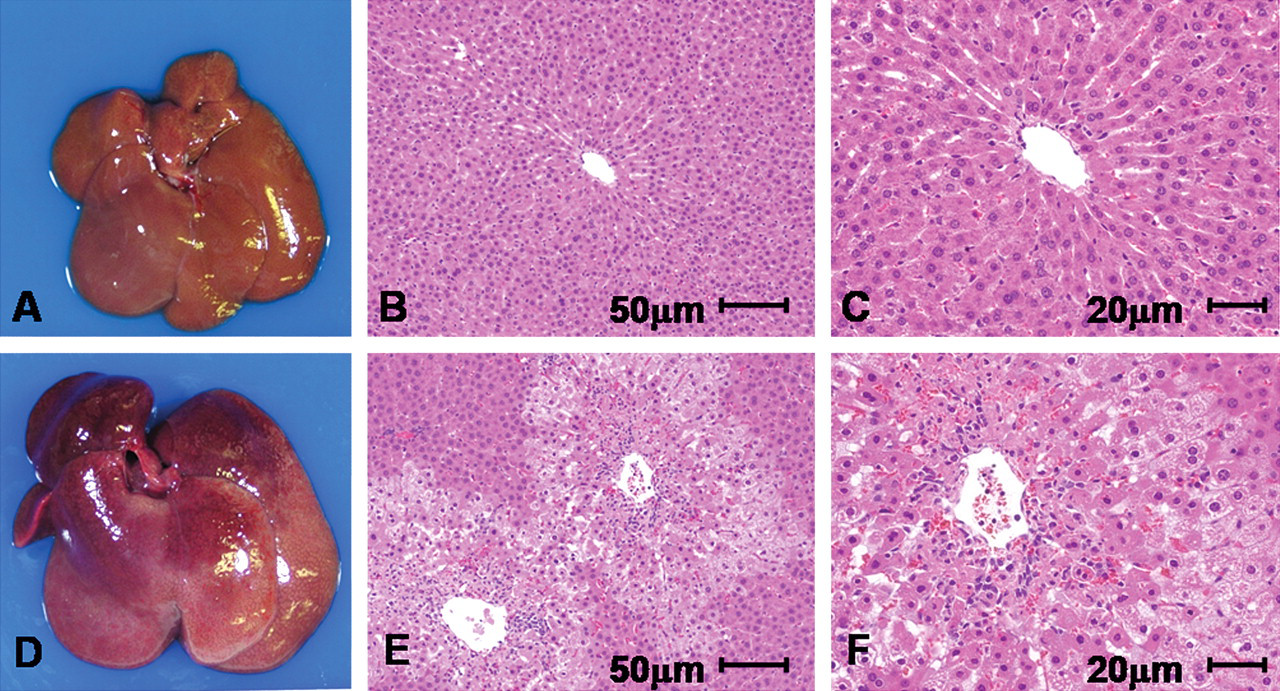
Morphological changes of liver in Tg and wild-type rats twenty-four hours after 500 mg/kg APAP exposure. (A, D) Macroscopic pictures of livers of Cx32ΔTg (A) and wild-type rats (D). Note gross enlargement with bleeding in the APAP-treated wild-type case. (B, C, E, F) Hematoxylin and eosin (H&E)–stained sections of livers of Cx32ΔTg (B and C) and wild-type rats (E and F). Centrilobular cell death, inflammatory infiltration, and bleeding are prominent in the wild-type rat case with APAP.
Liver weights and serum biochemistry after treatment with various doses of APAP.
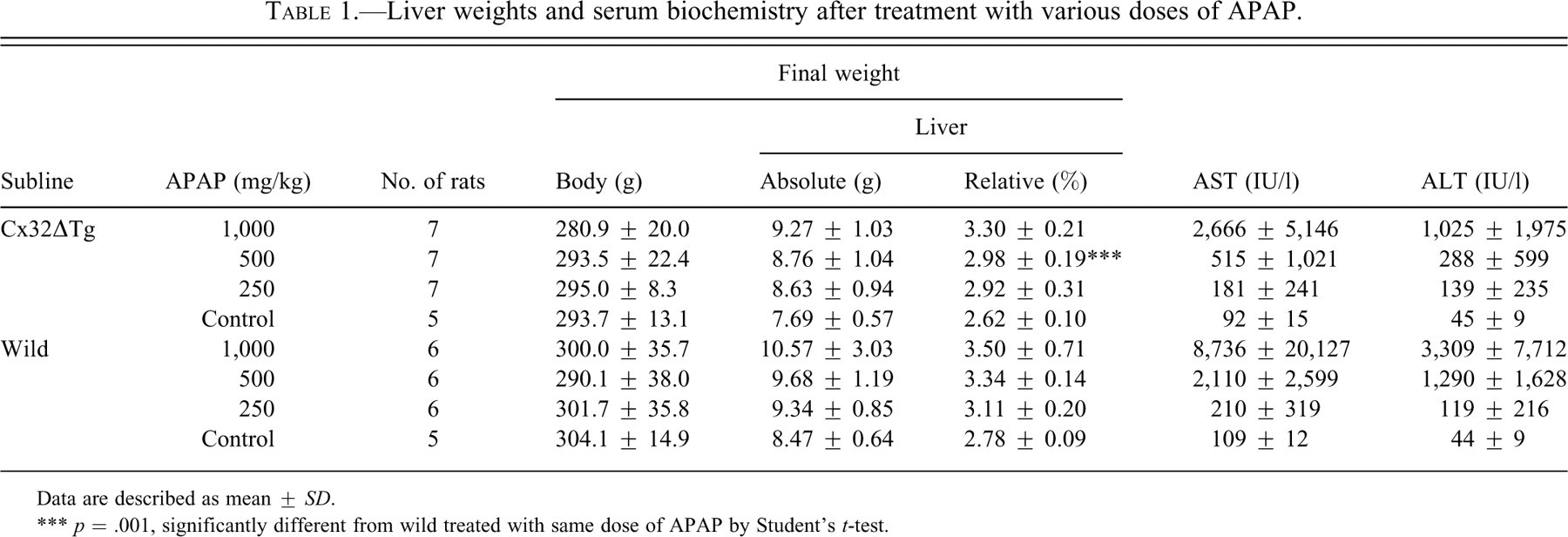
Data are described as mean ± SD.
*** p = .001, significantly different from wild treated with same dose of APAP by Student’s t-test.
Histological score of liver damage by APAP.
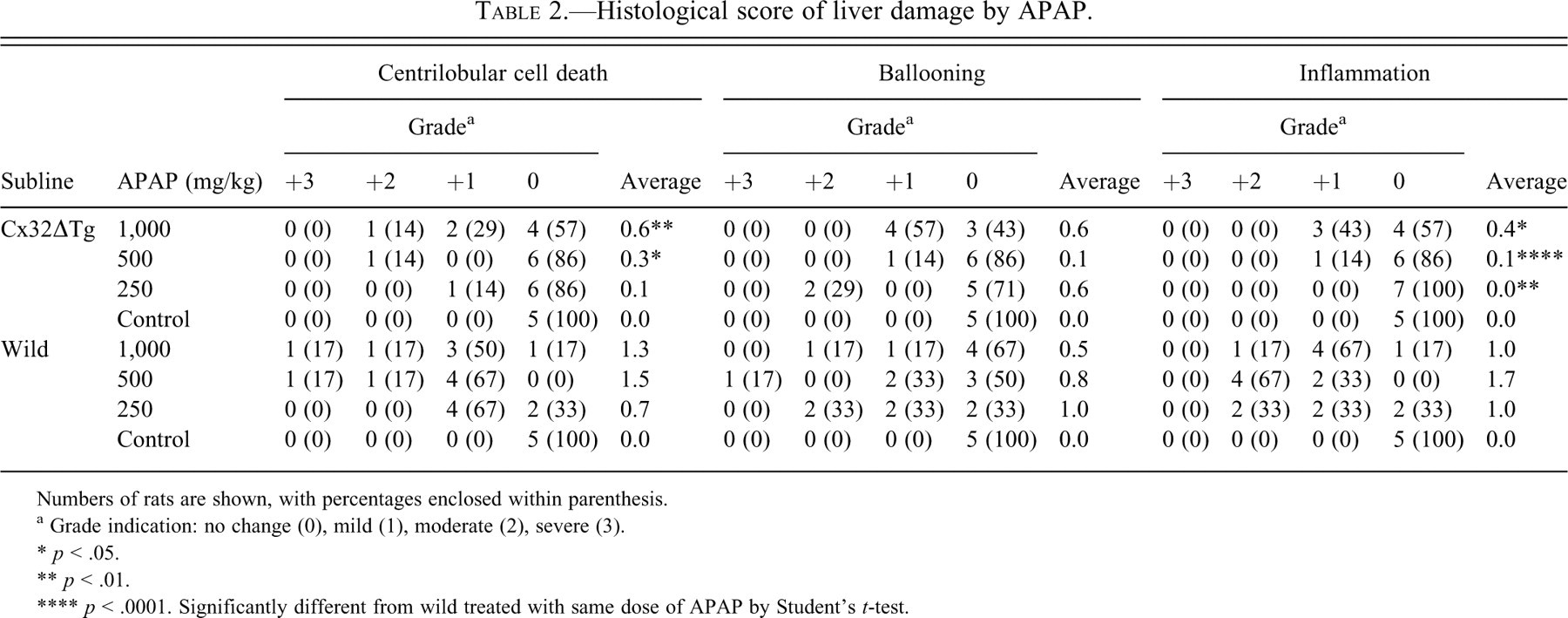
Numbers of rats are shown, with percentages enclosed within parenthesis.
a Grade indication: no change (0), mild (1), moderate (2), severe (3).
* p < .05.
** p < .01.
**** p < .0001. Significantly different from wild treated with same dose of APAP by Student’s t-test.
Because some chromatin condensed single cells were observed, as shown in Figure 2 (arrows), TUNEL assays were performed and positivity was found in such dead hepatocytes in the centrilobular zone. Additional immunohistochemical study revealed some TUNEL-positive cells to express cleaved caspase-3 (Figure 3 ). Expression of Cx43 is not observed in normal hepatocytes. However, some reports indicated Cx43 induction in apoptosis of rat bladder carcinoma and sarcoma cells (Krutovskikh, Piccoli, and Yamasaki 2002; Saito et al. 2007; Suzuki et al. 2005). Thus, we investigated whether Cx43 expression will be induced in apoptotic phase of hepatocytes, and expression of Cx32 was also examined. Cx32 protein was immunohistochemically localized at the cell membrane of normal hepatocytes, reflecting formation of GJIC in wild-type rats. In Cx32ΔTg rats, however, staining was very limited as previously reported (Asamoto et al. 2004) (Figure 4 ). In damaged hepatocytes of wild-type rats, Cx32 expression was presented in cytoplasm rather than cell membranes. On the other hand, Cx43 protein was induced in the cytoplasm of damaged hepatocytes. Furthermore, merged images for Cx32 and Cx43 immunofluorescence revealed coexpression of the two connexins in some damaged hepatocytes (Figure 4). Western blotting analysis further indicated that Cx43 protein expression was induced parallel to activation of caspase-3 in damaged livers (Figure 5 ).
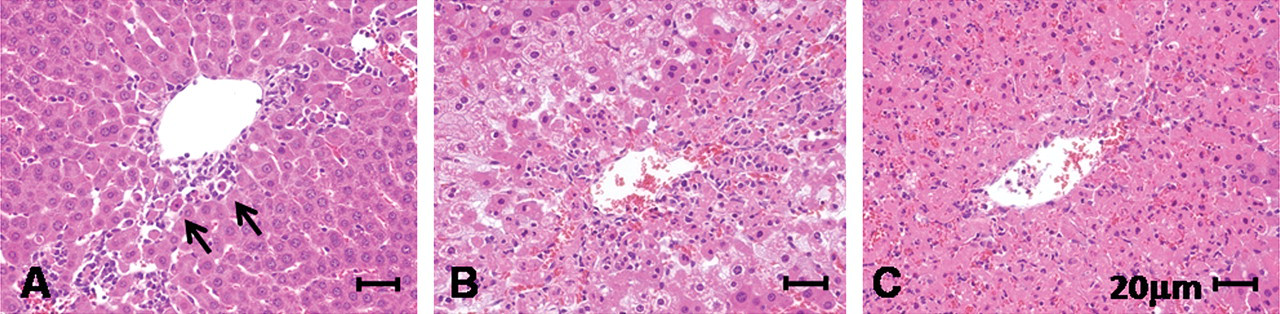
Histological scores for hepatotoxicity of APAP. The three pictures show typical histology of wild-type rat livers exposed to APAP. (A) 250 mg/kg APAP treatment; centrilobular cell death (+1), ballooning (0), and inflammation (+1). Arrows indicate single cell death. (B) 500 mg/kg APAP treatment; centrilobular cell death (+2), ballooning (+3), and inflammation (+2). (C) 500 mg/kg APAP treatment; centrilobular cell death (+3), ballooning (+1), and inflammation (+2).
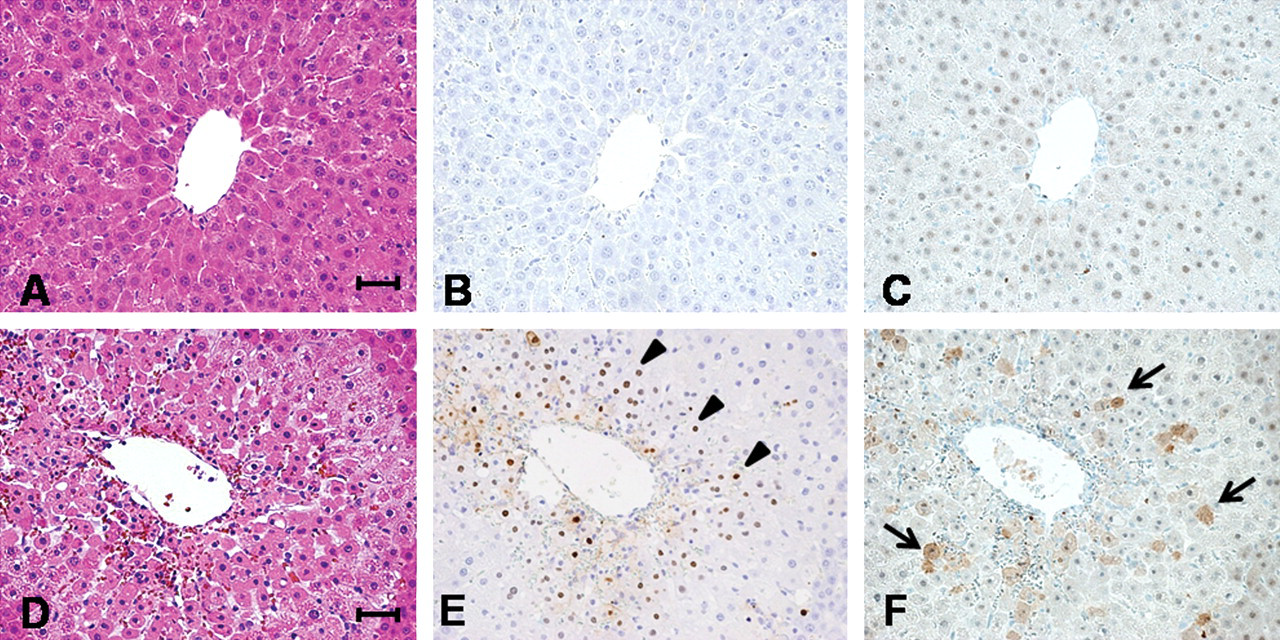
Analyses for apoptosis by TUNEL assay and immunohistochemistry for cleaved caspase-3. Upper pictures show liver histology of Cx32ΔTg, and lower show wild-type. Hematoxylin and eosin (H&E) staining sections (A, D) are presented at same position of TUNEL assay (B, E) and cleaved caspase-3 (C, F). TUNEL-positive cells were pointed by arrowheads, and arrows indicate positive staining for activated caspase-3 in cytoplasm.
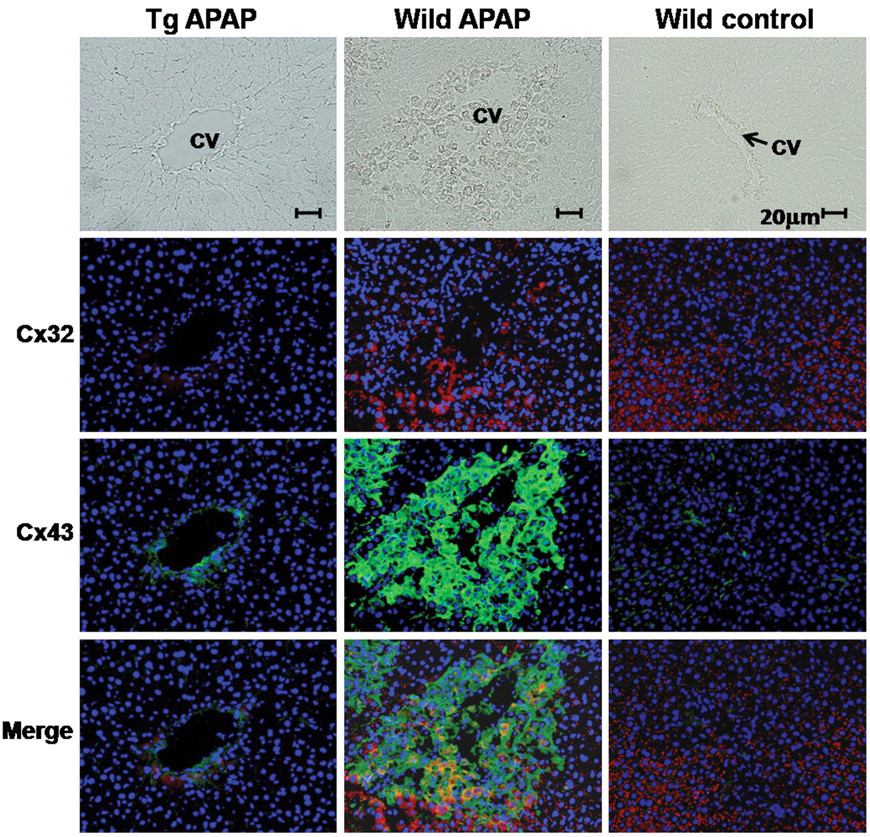
Immunohistochemical analyses for Cx32 and Cx43. Pictures in left and middle line indicate APAP-treated Cx32ΔTg and wild-type livers, and pictures in right line show control wild-type liver. Cx32 and Cx43 are visualized by using TRITC-labeled and FITC-labeled streptavidin under fluorescence microscopy. Lower pictures indicate combination of Cx32 and Cx43 staining. CV: central vein.
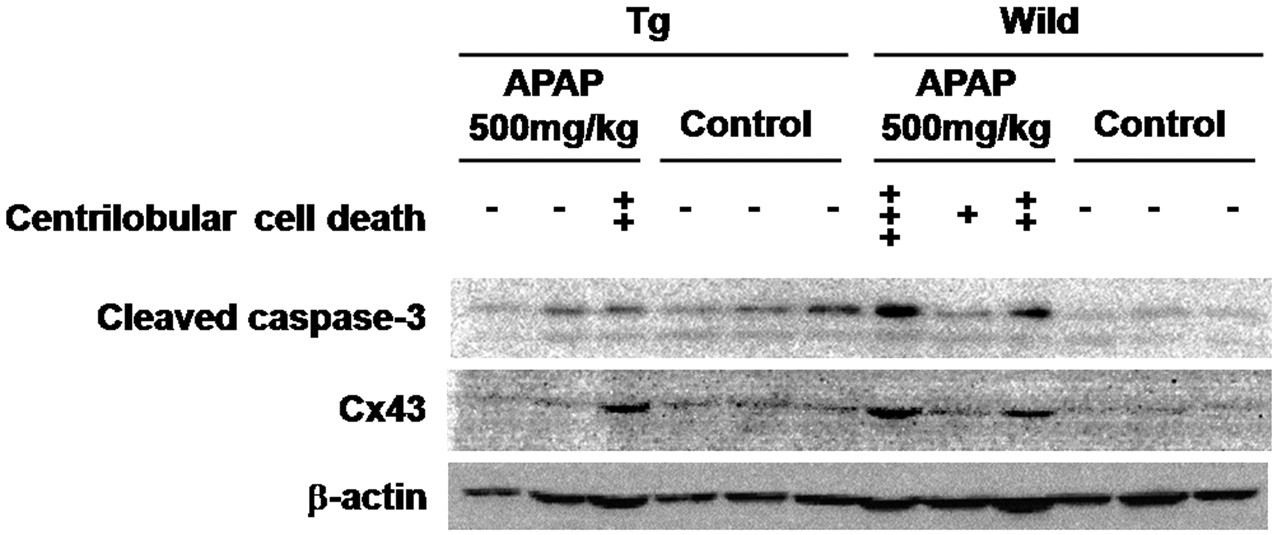
Western blot of APAP-exposed and control liver tissues. Lane 1 to 3: Cx32ΔTg livers exposed to 500 mg/kg of APAP. Lane 4 to 6: Cx32ΔTg livers as control. Lane 7 to 9: Wild-type livers exposed to 500 mg/kg of APAP. Lane 10 to 12: Wild-type livers as control. Scores of centrilobular cell deaths are indicated as described in the Materials and Methods section.
Discussion
The present study revealed hepatotoxicity of APAP in Cx32ΔTg rats to be much less prominent than in the wild-type (Tables 1 and 2; Figures 1 and 2). It is thus clear that GJIC participates in APAP-induced hepatotoxicity. Similar findings were observed in our previous study in which Cx32ΔTg rats were demonstrated to be resistant to liver injury induced by D-galactosamine and carbon tetrachloride (Asamoto et al. 2004). Metabolic factors may make important contributions to chemical-induced hepatotoxicity. However, we considered that they may not have been directly related to the hepatotoxicity in this model, because mRNA levels of cytochrome P450s, which play a critical role in the metabolism of many drugs including APAP, were found to not significantly differ between Cx32ΔTg and wild-type rats (data not shown).
Histological examination indicated that there was no clear difference in APAP-induced hepatocellular damages between 1,000 and 500 mg/kg in wild-type rats (Table 2). This finding suggests that a dose of 500 mg/kg is high enough to induce severe toxicity in a normal liver. In Cx32ΔTg, there was little influence of APAP at doses of 500 or 250 mg/kg, and only half of the Cx32ΔTg rats in the high-dose group (1,000 mg/kg) developed liver injury (Tables 1 and 2). Hepatotoxicity of such a high-dose APAP may not be modified by GJIC. In human cases, APAP-associated hepatotoxicity may occur not only with high doses but also with recommended use (Floren, Thesleff, and Nilsson 1987; Moling et al. 2006; Seirafi, Iten, and Hadengue 2007). Whether individual variability reflects any differences in gap junction activity remains to be clarified.
There have been many reports of APAP-induced cell death in the liver. For example, apoptosis has been described with exposure of APAP in mouse hepatocytes both in vitro and in vivo (El-Hassan et al. 2003; Ray et al. 1996). On the other hand, another report proposed that necrosis is the principal mechanism of liver-cell death by APAP, with apoptosis found in only very few mice in vivo (Gujral et al. 2002). According to previous reports, apoptosis and necrosis frequently coexist in liver pathology, and the actual mode of cell death may be regulated by the particular circumstances. In the present study, the damaged hepatocytes shrank with chromatin condensation. TUNEL assays also indicated marked apoptotic signaling in APAP-exposed livers, with activated caspase-3 expression in TUNEL-positive cells (Figure 3), and the expression of cleaved caspase-3 correlated with degree of hepatotoxicity (Figure 5). These results suggest that apoptosis through caspase-3 is an important mechanism for hepatocyte death occurring as a consequence of APAP toxicity.
Immunohistochemistry here suggested that Cx32 expression was lost from cell membranes of apoptotic hepatocytes, while another connexin protein, Cx43, was induced in the cytoplasm of the cells (Figure 4), and Cx43 protein expression was related to activation of caspase-3 in damaged livers (Figure 5). Given the previous report of Cx43 expression in apoptotic rat bladder carcinoma cells (Krutovskikh, Piccoli, and Yamasaki 2002), this form may contribute to apoptosis in both normal and cancerous cells. As far as we know, this is the first evidence of roles of Cx43 in apoptosis in normal cells. The role of Cx43 in apoptosis has yet to be fully understood. A previous study indicated the possibility that Ca2+ ions are messenger molecules that pass through Cx43-formed gap junctions to kill cells in vitro (Krutovskikh, Piccoli, and Yamasaki 2002). In the present in vivo study, because Cx43 protein was diffusely located in cytoplasm, it may be involved with apoptosis as a GJIC-independent molecule.
Cx43 protein was also found to be present in dead hepatocytes of APAP-treated Cx32ΔTg as in wild-type rats, and its expression level depended on the degree of hepatotoxicity as illustrated in Figure 5 (immunohistochemical data not shown). This finding suggests that apoptosis mediated by Cx43 induction may be a principal process of APAP-induced hepatic cell death, regulated by Cx32-formed GJIC. Thus, Cx43 expression itself could be controlled by messengers passing through gap junctions, like Ca2+ and ATP, although further studies are necessary to identify detailed mechanisms. Our previous studies using the Cx32ΔTg rat model suggests that disruption of GJIC results in resistance to hepatotoxicity and enhancement of susceptibility for hepatocarcinogenesis (Asamoto et al. 2004; Hokaiwado et al. 2007, 2005). From our present results, we propose that under decreased GJIC conditions, APAP-injured hepatocytes fail to be removed by apoptosis, and these cells could be a population with a predisposition for carcinogenic alteration.
In conclusion, acute APAP-induced hepatotoxicity depends on Cx32-associated gap junctional functions in the liver. In the wild-type case, APAP-injured hepatocytes may undergo apoptotic death in some way linked to Cx43 expression.
Footnotes
Acknowledgments
The authors thank Dr. Malcolm A. Moore for his kind linguistic advice during preparation of this article.