
Abstract
If p53 is essential to eliminate damaged spermatogenic cells, then mutagen exposure in the absence of p53 would increase sperm containing damaged DNA. p53 knockout (−/−, NULL) and wild-type (+/+, WT) mice (five/group) were exposed to ethylnitrosourea (ENU) or cyclophosphamide (CP). In phase I, mice were exposed by gavage to 0 or 60 mg/kg/day ENU or CP for four days and examined on test day (TD) 4, and in phase II, mice were exposed to 0, 6, 20, or 60 mg/kg/day ENU or CP for four days and evaluated on TD 36 when exposed spermatocytes matured. In phase I, mutagens were not directly cytotoxic to mature sperm. In phase II, WT mice were more sensitive to decreases in reproductive organ weights, whereas both genotypes had decreased sperm counts. Testicular histology revealed similar CP responses, but genotype-specific ENU responses (WT mice had depletion of elongating spermatids; NULL mice had late-stage spermatocyte/early stage spermatid loss). Ethylnitrosourea increased DNA strand breaks in WT mice. Thus, mice responded similarly to CP, suggesting a primarily p53-independent response, whereas the ENU response differed by zygosity, suggesting a role for p53. As DNA damage increased at higher ENU doses, compensatory repair pathways may operate in NULL mice.
Introduction
Trp53 (p53) is a transcription factor that up-regulates, transactivates, and represses the expression of genes and proteins responsible for cell cycle arrest and apoptosis (Oren 2003; Resnick et al. 2005). Mice deficient in p53 protein (p53−/−) have a greater tendency to develop spontaneous tumors. Aside from somatic cells, p53 plays a role in maintaining genetic integrity of the gametes. p53 is highly expressed in the testes (Beumer et al. 1998; Chandrasekaran and Richburg 2005); it is present in spermatogonia (Beumer et al. 1998) and primary spermatocytes during the pachytene (Schwartz et al. 1993) or preleptotene to early pachytene stages (Almon et al. 1993; Schwartz et al. 1993; Sjöblom and Lähdetie 1996), when chromosomal pairing, recombination, and DNA repair occur (Alcivar et al. 1992). Expression at these stages of spermatogenesis suggests that p53 plays a role in meiosis, a point substantiated by the presence of multinucleated giant cells in the seminiferous tubules of p53 knockout mice (Rotter et al. 1993). If DNA damage is severe, p53 plays a role in triggering germ cell apoptosis, although p53-independent pathways also exist (Beumer et al. 1998). Apoptosis is critical for the maintenance of germ cell DNA and appropriate germ cell numbers.
In our original study (Marty et al. 1999), we hypothesized that the absence of adequate concentrations of p53 would result in aberrant spermatogenesis or sperm containing damaged DNA. However, there were no qualitative differences in the incidence of sperm shape abnormalities and sperm counts, regardless of p53 zygosity (nullizygous [−/−, NULL], heterozygous [+/−], wild-type [+/+, WT], or inbred C57B16 mice). In contrast, in both NULL and heterozygous p53 knockout mice, there were fewer strand breaks in sperm DNA, suggesting that the type and/or number of DNA-protein or DNA-DNA cross-links in p53 (−/−) and (−/+) mice differ from their WT (+/+) counterparts. Given these data, we hypothesized that the background incidence of genetic damage in heterozygous and homozygous p53 knockout mice was not sufficiently severe to induce marked changes in the sperm end points measured.
It is possible that the role of p53 in spermatogenesis was underestimated in our previous study because of the lack of an environmental stressor(s) and insufficient DNA damage. DNA strand breaks are critical for the elevation of p53 protein levels (Nelson and Kastan 1994), and in normal cells, there is little p53 until exposure to DNA-damaging agents. Agents inducing rapid DNA double-strand breaks have been associated with elevations in p53 protein (Nelson and Kastan 1994). Irradiation, which induces DNA double-strand breaks, has been demonstrated to increase p53 expression in spermatocytes (Sjöblom and Lähdetie 1996) and spermatogonia (Beumer et al. 1998). Thus, the necessity of p53 expression during spermatogenesis may be linked to the type of DNA alterations present.
Given these previous results, we hypothesized that if DNA damage is a prerequisite for elevated p53 expression and the specific type of DNA lesion (e.g., DNA double-strand breaks) plays a role in p53-mediated genomic repair, then p53 knockout mice exposed to clastogenic agents will exhibit sperm parameters that are significantly different from similarly treated WT mice. For this study, ethylnitrosourea (ENU) and cyclophosphamide (CP) were selected.
Numerous studies have shown that ENU alters spermatogenesis. Ficsor et al. (1984) reported altered mouse sperm morphology forty-nine days after a single ip treatment with 100 mg/kg ENU. Skare and Schrotel (1984) have shown DNA single-strand breaks in rat testicular DNA following ip injection of 85.2 mg/kg ENU. Oral administration of ENU (four daily doses of 12.5, 50, or 100 mg/kg) increased the mutation frequencies in spermatogenic cells at a variety of stages (Katoh, Horiya, and Valdivia 1997), a finding substantiated by Ashby et al. (1997). Ethylnitrosourea produces DNA single-strand breaks and alkali-labile lesions by alkylating DNA, particularly at O6-guanine, and forming phosphotriesters. During the repair process, these alkylation sites are depurinated, and these apurinic sites are transformed to DNA single-strand breaks, a finding demonstrated in several in vitro assays (Abbondandolo et al. 1982; Vitelli et al. 1989), including the comet assay (Henderson et al. 1998; Muller et al. 1997). Ethylnitrosourea, operating in a dose- and time-dependent fashion, also has been implicated in the production of DNA double-strand breaks through the induction of gammaH2AX foci in a human amnion FL cell line (Zhou et al. 2006).
Similarly, CP treatment has been shown to induce sperm morphology abnormalities (Chauhan et al. 2000; Ribeiro et al. 1987; Salomaa, Donner, and Norppa 1985). Although sperm abnormalities were increased in these studies, there is conflicting evidence on CP-induced alterations in sperm number. In the study by Salomaa, Donner, and Norppa (1985), sperm counts were not significantly affected thirty-five days after treatment with 40 or 80 mg/kg CP, whereas Chauhan et al. (2000) reported effects on sperm numbers one month after treatment with 20 mg/kg CP. Male Sprague-Dawley rats had transient effects on relative testicular and cauda epididymal sperm counts (numbers/g tissue) after three and six weeks, respectively, of oral exposure to 5.1 or 6.8 mg/kg/day CP (Trasler, Hales, and Robaire 1987). A review of the literature indicates that there are numerous modes of genotoxic activity for CP. Cyclophosphamide is activated by hepatic mixed-function oxidases. Its metabolites can induce genetic damage in germ cells and are considered clastogenic (Kamiguchi and Tateno 2002). When used in a variety of genotoxicity assays, CP has been shown to form DNA adducts, as well as to stimulate unscheduled DNA synthesis, sister chromatid exchange, base-pair substitutions, gene mutations, and chromosomal aberrations (Anderson et al. 1995). Cyclophosphamide (6 or 50–100 mg/kg/day) caused dose-dependent increases in DNA strand breaks in spermatozoa collected from Sprague-Dawley rats fourteen to twenty-eight days after four daily doses (Codrington, Hales, and Robaire 2004). An increased frequency of aberrant cells was observed using 20 mg/kg CP in the mouse bone marrow chromosomal aberration test (Chauhan et al. 2000). Although one reference reports that CP is negative for aneuploidy (Anderson et al. 1995), a second study reports that CP causes metaphase II hyperploidy when injected into mice (Allen et al. 1986). At high dose levels, CP reportedly affects cell division (Anderson et al. 1995). Cyclophosphamide is routinely used in a number of laboratories as a positive control agent for the induction of chromosomal damage in the bone marrow cells of both rats and mice.
The purpose of this study was to determine whether mutagen-induced DNA lesions could trigger p53-dependent genomic repair. It was hypothesized that if p53 is essential for normal spermatogenesis and elimination of spermatogenic cells with DNA damage, then treatment of p53 NULL mice with mutagens would result in aberrant spermatogenesis or retention of sperm with damaged DNA owing to the absence of adequate concentrations of p53. To test this hypothesis, p53 wild-type (+/+, WT) and nullizygous (−/−, NULL) mice were exposed to CP or ENU by oral gavage for four days. In phase I, WT or NULL mice exposed to 60 mg/kg/day CP or 60 mg/kg/day ENU were compared with control mice of their respective zygosity on test day (TD) 4 (approximately four hours after the final treatment) to assess acute effects on mature sperm. In phase II, a more complete examination of sperm parameters was conducted in mice exposed to 6, 20, or 60 mg/kg/day ENU or 6, 20, or 60 mg/kg/day CP at thirty-six days post-initiation of treatment (thirty-two days after cessation of treatment). Test day 36 was selected for phase II examinations to allow mutagen-exposed spermatocytes to mature into spermatozoa. This research project was designed to examine the sensitivity and specificity of the p53 transgenic mouse model for various types of DNA-damaging agents and to better characterize the DNA repair mechanisms intrinsic to p53 knockout mice.
Materials and Methods
Mice
Nullizygous p53 knockout (p53−/−; model # P53N4-M; B6.129-Trp53tm1Brd N4) and WT (p53+/+; model # P53N5-W; B6.129-Trp53tm1Brd N5) mice were purchased from Taconic (Germantown, NY, USA). As a result of the introduction of a null mutation by homologous recombination, the NULL mice lack a functional p53 allele (Donehower et al. 1992). The absence of p53 protein in the NULL mice was confirmed by the supplier. Mice were approximately seven weeks of age upon arrival and were maintained in the laboratory for at least seven days prior to the initiation of treatment. The laboratory was fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International). In the animal rooms, the temperature was maintained at 22 ± 3°C, the relative humidity was 40–70%, and the photoperiod was 12 hr. Animals were housed two or three to a cage and given feed (PMI Certified Rodent Lab Diet #5002; PMI Nutrition International, St. Louis, MO, USA) and municipal tap water ad libitum. Mice were randomized into treatment groups (five mice/genotype/group) using a computer program designed to yield approximately equal mean body weights and variances across all groups. Once placed on study, animals were housed individually.
Study Design
This study was designed in two phases; phase I involved an evaluation of the acute effects of high-dose ENU and CP treatment, and phase II examined persistent dose-related effects of ENU or CP on spermatogenesis. For both phases, mice from each genotype received four consecutive daily doses of ENU, CP, or vehicle (phosphate-buffered saline, PBS) by oral gavage. Cyclophosphamide and ENU were supplied by Sigma (St. Louis, MO, USA). The study design is illustrated in Figure 1.
An outline of the experimental study design. CP = cyclophosphamice; ENU = ethylnitrosourea.
For phase I, fifteen p53+/+ and fifteen p53−/− male mice (five per dose level) were dosed by oral gavage for four consecutive days with 60 mg/kg/day ENU, 60 mg/kg/day CP, or a vehicle (phosphate-buffered saline [PBS]) control. Approximately four hours after treatment on TD 4, animals were humanely euthanized for the evaluation of epididymal sperm counts and DNA strand breaks in the spermatozoa using the comet assay (see below).
In phase II, reproductive organs and germ cells were evaluated thirty-six days after the first treatment with ENU or CP to evaluate organ level effects, as well as the degree of unrepaired DNA damage remaining in sperm that have matured from exposed spermatocytes (Foster et al. 1983; Rugh 1968). The thirty-six-day interval was selected to allow exposed spermatocytes to develop into spermatozoa, and for the spermatozoa to reach the epididymis for collection (Shelby et al. 1986). Basically, WT p53+/+ (thirty-five mice) and NULL p53−/− (thirty-five mice) were divided into fourteen groups (five WT or five NULL mice/group) and administered ENU at three dose levels (6, 20, or 60 mg/kg/day), CP at three dose levels (6, 20, or 60 mg/kg/day), or vehicle control solution (PBS) by oral gavage for four consecutive days. On TD 36, mice were euthanized and testes and epididymides were removed and weighed. In most cases, the right testis was preserved for histopathological examination and the right cauda epididymis was used for sperm counts, whereas sperm were collected from the left cauda epididymis for evaluation of DNA strand breaks (comet assay) and apoptosis (DNA diffusion assay). The exceptions to this assignment plan were in the case of WT mice that exhibited unilateral enlarged caput epididymis, which was unrelated to mutagen treatment. Two WT mice had enlarged left caput epididymis; therefore, in these animals, the right side (grossly normal) tissues were used for the comet and apoptosis assays. Preference was given to the comet and apoptosis samples, as those assays offered a better assessment of DNA integrity. Sperm counts were excluded for animals with enlarged caput epididymides (see below).
Dosing
The ENU dosing solution was prepared by dissolving ENU in PBS buffered to approximately pH 6.0. The ENU dose solution was prepared each day immediately prior to dosing. This ENU dosing regimen has been shown to increase mutation frequencies in spermatogenic cells at a variety of stages (Katoh, Horiya, and Valdivia 1997).
The CP dose solution was prepared by dissolving CP in distilled water. The solution was prepared on TD 1 and used on the three subsequent days of dosing. The CP dosing paradigm was adapted from studies by Salomaa, Donner, and Norppa (1985); Ribeiro et al. (1987); and Chauhan et al. (2000), in which CP treatment induced sperm abnormalities.
Both the ENU and CP dosing solutions were administered at a dose volume of 15 mL/kg/day. The control animals received the same volume of PBS buffered to pH 6. Although a separate distilled water control group (vehicle for CP exposures) was not included in the study, it is unlikely that either the PBS or distilled water altered measured parameters. Results for control animals in the current study were qualitatively similar to previously reported studies comparing p53 WT and NULL mice, for example, greater testes weights and epididymal sperm counts in NULL mice compared with WT mice (Beumer et al. 1998; van Buul et al. 2002), and slightly lower mean spontaneous sperm apoptosis in p53−/− vs. p53+/+ control mice (Yin et al. 1998).
Phases I and II: Clinical Observations and Body Weights
Both phase I and phase II animals were subjected to daily handheld clinical examinations after test material administration on TDs 1–4. In addition, all animals were given daily cageside examinations throughout the course of the study. Phase II animals also were given weekly handheld clinical examinations to evaluate their general health status after the four-day dosing period. Mice were weighed pre-exposure and on TDs 1, 2, 3, 4, 8, 15, 22, 29, and 36.
Phases I and II: Necropsy
Animals submitted alive for necropsy were anesthetized by the inhalation of carbon dioxide. The animals were euthanized by cervical dislocation, terminal body weights were collected, the skin was reflected from the carcass, and the thoracic and abdominal cavities were opened and examined. For TD 4 animals (phase I), the epididymides were removed and trimmed. A small hole was poked in the left cauda epididymis with a 27-gauge needle. A small sample of sperm was expressed into a tube containing RPMI 1640 with 10% fetal bovine serum and 1% bovine serum albumin (Fraction V). Sperm were allowed to swim out of the cauda for five minutes, then the samples were immediately placed on ice (Marty et al. 1999). The cauda from the right epididymis was weighed and quick-frozen in liquid nitrogen for subsequent sperm counts. All other tissues and the carcasses were discarded.
For phase II animals, necropsies were similar to phase I animals. The epididymides and testes were removed, trimmed, and weighed. The right testis was placed in Bouin’s fixative for subsequent histological examination. The right cauda epididymis was saved for sperm counts. Sperm was collected from the left cauda epididymis as described above for the comet and apoptosis assays, as described above.
Testes fixed in Bouin’s solution were embedded in paraffin, sectioned 5 µm thick, stained with periodic acid-Schiff (PAS) stain, and examined by a veterinary pathologist. A qualitative evaluation of the testis included examination for retained spermatids, missing germ cell layers or types, multinucleated giant cells, and sloughing of spermatogenic cells into the lumen.
Phases I and II: Epididymal Sperm Counts
The right epididymis was frozen at –80°C for determination of cauda epididymal sperm reserves. The thawed epididymides were minced thoroughly in a solution containing 150 mM NaCl, 0.05% Triton X-100 (v/v) and 0.25 mM merthiolate (STM medium). The resulting cell suspension was allowed to stand for fifteen minutes for sperm to dissociate from the epididymal tissue. An aliquot of this suspension was counted using a hemacytometer. If counts differed markedly between the two sides of the hemacytometer (i.e., >15% of the mean), the sample was recounted. Total sperm count per epididymis and per gram of epididymal tissue was calculated. From these individual values, a group mean was calculated.
Phase I and II: Comet Assay
The epididymal sperm sample collected at necropsy was overlaid with mineral oil to fill the remaining head space in the tube and shipped on ice to the University of Washington (Seattle, WA, USA) for analysis of DNA strand breaks and apoptosis using the comet and DNA diffusion assays, respectively. The protocol used for the comet assay has been described previously (Marty et al. 1999), with some modifications. Basically, sperm cells were pelleted and resuspended in PBS to yield 1000 cells/μL. An aliquot of the sperm cell suspension (10 µL) was mixed well with 50 µL 0.7% 3:1 high-resolution agarose (Amresco, Solon, OH, USA) in PBS and layered onto a clear-window, partially frosted, agarose-coated slide (MGE slide, Erie Scientific, Portsmouth, NH, USA). After removing the coverglass, another layer of 200 μL agarose was layered on top of the agarose layer with cells. This thicker third layer was added to prevent DNA from escaping during lysis and electrophoresis. After removing coverglasses, slides with microgels were immersed for two hours at 4°C in cold lysing solution containing 1.25 M NaCl, 0.01% sodium N-lauroyl sarcosinate, 50 mM tetrasodium ethylenediaminetetraacetic acid (EDTA), 100 mM Tris (pH 10), and 1 mg/mL reduced glutathione (crystalline free acid). Slides were incubated for two hours in a prewarmed (37°C) lysing solution containing 1.25 M NaCl, 5 mM tetrasodium EDTA, 5 mM Tris (pH 10), and 0.5 mg/mL of DNAse-free proteinase K (Amresco, Solon, OH, USA), and then placed on a horizontal electrophoretic unit (Ellard Instrumentation, Monroe, WA, USA) that was modified to allow electrical input from a power supply to both ends of an electrode. The unit was filled with 1 L of 500 mM NaCl, 100 mM Tris HCl (pH 9), and 1 mM EDTA. Slides with microgels were allowed to equilibrate for twenty minutes and electrophoresed for twenty min at 12 volts and 250 milliamperes while the solution was recirculated at ~100 mL/min. The electrophoretic unit was designed to have uniform electrical fields, thereby minimizing slide-to-slide variation in DNA migration. Slides for neutralization and DNA precipitation were immersed in freshly prepared 1 M ammonium acetate in 95% ethanol for 15 min and then 20 mM Tris (pH 7.4) in 50% ethanol with 1 mg/mL of spermine for 10 minutes, and the step was repeated once more. Slides were immersed in 75% ethanol containing 20 mM Tris (pH 7.4) for ten minutes. This step was repeated twice more, and the slides were air-dried. One slide at a time was stained with 50 μL 0.25 µM of YOYO-1 in 0.5% sucrose and 2.5% dimethyl sulfoxide (DMSO). Fifty images from each slide were captured using VisComet image analysis software Version 1.54 (Impulse Bildanalyse GmbH, Gilching, Germany January 2001) and CCD camera GW525x (Genwac Inc., Orangeburg, NY, USA) attached to a DMLB epifluorescence microscope (Leica, Germany) with excitation at 490, dichroic at 500, and emission at 515 nanometers. It is our laboratory’s experience that use of a specially designed electrophoretic unit during electrophoresis results in uniform electrical fields and insignificant slide-to-slide variation in DNA migration; thus, it was considered unnecessary to use two slides from each animal. Comet Extent, which included diameter of head (nucleus) and tail length (migrated DNA, up to last pixel of DNA) in pixels, was used as an index of DNA strand breaks. All samples were scored blind to treatment group.
Phase II: Apoptosis
The DNA diffusion assay was performed as previously described (Singh, Muller, and Berger 2003). Basically, approximately 100,000 sperm cells were mixed with 50 µL of 1% agarose 3:1, and this mixture was pipetted onto an MGE slide that was already coated with a dry layer of agarose 3:1. The agarose cell mixture was immediately covered with a 24 × 50 mm2 #1 cover glass (Corning Glass Works, Corning, NY, USA) and cooled in a steel tray on ice for one minute. The cover glass was removed, and 200 µL of 2% SFR agarose (Amresco, Solon, OH, USA) solution was layered as before to make a third layer. Use of 2% SFR agarose is essential for controlling too much diffusion of DNA from apoptotic cells in agarose. After keeping slides for two minutes on ice, cover glasses were removed and the slides were immersed and maintained for ten minutes in a freshly made and cold lysing solution (1.25 M NaCl, 1 mM tetrasodium EDTA, 5 mM Tris, 0.01% sodium lauroyl sarcosine, 0.2% DMSO, and 300 mM NaOH). Slides for neutralization and DNA precipitation were immersed in freshly prepared 1 M ammonium acetate in 95% ethanol for 15 min and then 20 mM Tris (pH 7.4) in 50% ethanol with 1 mg/mL of spermine for ten minutes, and the step was repeated once more. Slides were then immersed in 75% ethanol containing 20 mM Tris (pH 7.4) for ten minutes. This step was repeated twice more, and the slides were air-dried. One slide at a time was stained with 50 μL 0.25 µM of YOYO-1 in 0.5% sucrose and 2.5% DMSO. The percentage of cells with diffuse DNA and a hazy outline (signs of apoptosis) were calculated from a total of 1,000 cells. All samples were scored blind with respect to genotype of the donor animal and treatment group.
Statistics
Experiments in WT and NULL mice were run separately; therefore, data also were analyzed separately within zygosities (WT or NULL). Descriptive statistics (means and standard deviations) of body weights, body weight gains, organ weights, and sperm counts were calculated. Group means were evaluated by Bartlett’s test for homogeneity of variance. Based on the outcome of this test, a parametric (n = 5) or nonparametric (n < 5) analysis of variance (ANOVA) followed by Dunnett’s test or Wilcoxon rank-sum test was performed. Apoptosis data and comet assay data, which measure DNA migration, were subjected to a Student-Newman-Keuls test in phase I (single dose levels), whereas in phase II, these data were analyzed using a one-way ANOVA with Dunnett’s test to compare the vehicle and treatment groups during post hoc comparisons. In all cases, the critical level for statistical significance was set a priori at α = .05. Because the WT and NULL experiments were conducted separately, this study was not designed to compare baseline values between untreated WT and NULL mice, but rather to compare the treated responses to their respective control (WT or NULL). Some observations on the relative values of the control groups are mentioned to show that the results in p53 NULL mice are consistent with previously reported findings.
Results
Phase I (TD 4)
Initial responses to ENU and CP (each at 0 or 60 mg/kg/day) were compared qualitatively between male WT mice (p53+/+) and knockout mice (p53−/−; NULL) on TD 4 after four days of treatment. There were no treatment-related effects on clinical observation. In phase I, WT mice treated with 60 mg/kg/day ENU lost weight by TD 4 relative to pre-exposure (phase I data not shown). All other groups either maintained their body weight or gained weight over the four-day treatment period. On TD 4 (phase I), there were no treatment effects on gross observation at necropsy, absolute cauda epididymal weights, or epididymal sperm counts. In the comet assay, sperm from WT mice on TD 4 had slightly increased DNA migration length with mutagen treatment, suggesting a slight increase in DNA strand breaks. With NULL mice, DNA from ENU- and CP-treated mice migrated a shorter distance relative to NULL controls, suggesting fewer DNA strand breaks or the presence of DNA-DNA or DNA-protein cross-links. Data from phase I are included online as supplemental data, Tables S1–3.
Phase II (TD 36)
Clinical Observations, Body Weights, and Gross Observations at Necropsy
Following administration of CP and ENU on TD 1–4, treated spermatocytes were allowed to mature to determine whether repair occurred between the initial insult to these spermatocytes in the testis and the appearance of these cells as mature sperm in the epididymis (phase II end points examined on TD 36). There were no treatment-related clinical observations in phase II animals. With both zygosities, mice given 20 or 60 mg/kg/day CP or 20 (NULL only) or 60 mg/kg/day ENU lost weight by TD 4 relative to TD 1 (Figure 2A, 2B). Generally, these body weight losses were minimal (<2%), although the WT 60 mg/kg/day ENU group lost 6%. All treatment groups, regardless of zygosity, gained weight prior to necropsy on TD 36.
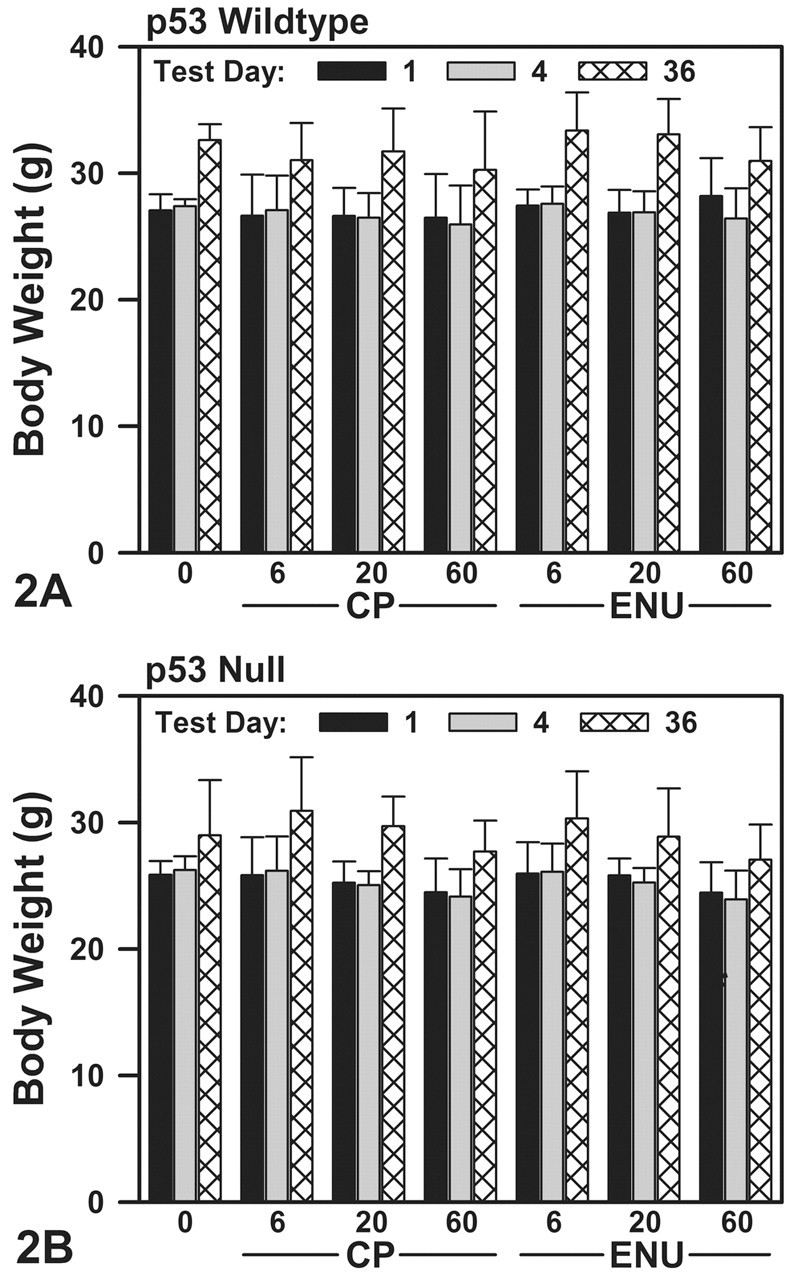
Body weights on TD 1, 4, and 36 in WT (A) and NULL (B) mice (mean + SD). WT mice treated with 60 mg/kg/day ENU weighed 6% less than controls at the conclusion of the treatment period on TD 4. All mice gained weight prior to necropsy on TD 36 (n = 5/group). CP = cyclophosphamice; ENU = ethylnitrosourea.
At necropsy, four WT mice had unilateral enlarged caput epididymides (one in the 6 mg/kg/day CP group, one in the 20 mg/kg/day CP group, and two in the 6 mg/kg/day ENU group). This observation was not seen in the high-dose groups, making its relationship to treatment questionable. Furthermore, with caput enlargement, epididymal weights were increased 48–125% relative to the mean weights in their respective dose groups, a result that conflicts with decreases in epididymal weights by all doses of ENU and the high dose of CP. Therefore, whole and cauda epididymal weights and epididymal sperm counts were excluded from analysis for these animals. For sperm counts, relative sperm counts were considered the primary parameter of interest. In addition, counts may have been influenced by altered fluid dynamics as opposed to direct effects of mutagen treatment. Histological examinations did not reveal corresponding testicular damage in these animals. Overall, the body weights and sperm counts from WT animals used in this study were consistent with data reported for the C57B16 strain of mice at similar ages (Chapin et al. 1993b; p53 NULL mice from Taconic were derived on strain 129 and back-crossed to C57B16).
Organ Weights
Data presented in Figure 3 are absolute testis and epididymal weights, which are conserved in the presence of minimal-to-moderate body weight changes (Chapin et al. 1993).
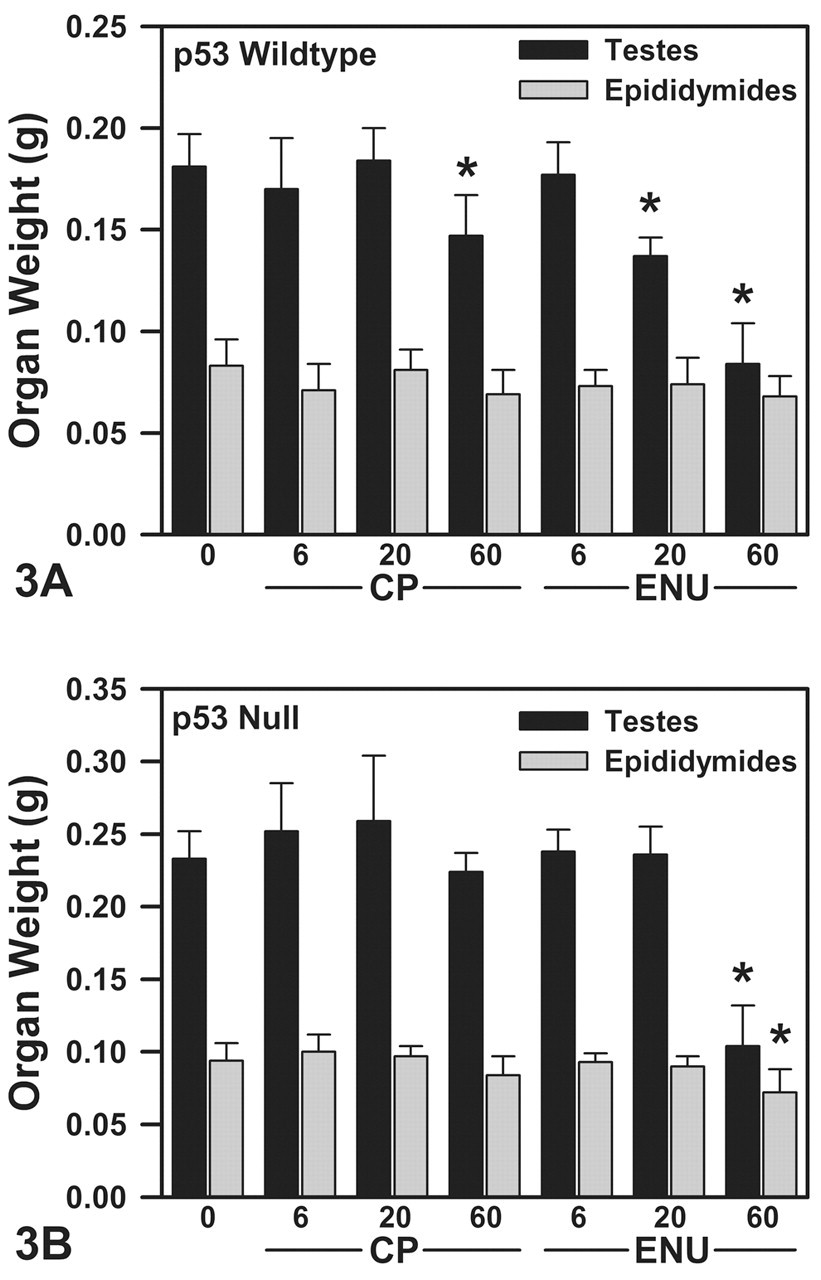
Reproductive organ weights on TD 36 in WT (A) and NULL (B) mice. (A) Cyclophosphamice (CP) (60 mg/kg/day) and ethylnitrosourea (ENU) (20 and 60 mg/kg/day) decreased paired testis weights in WT mice. Epididymal weights were decreased to a degree similar to testis weights in mice given high-dose CP or ENU, but these differences were not statistically significant. (B) In NULL mice, CP did not significantly decrease reproductive organ weights. With 60 mg/kg/day ENU, testis and epididymal weights were significantly decreased. Reproductive organ weights are expressed as means + SD. Asterisks denote values statistically different from controls by Dunnett’s test at α = .05 (n = three to five/group in WT mice and five/group in NULL mice).
WT ENU
Ethylnitrosourea significantly decreased testis weight in a dose-related manner at 20 and 60 mg/kg/day (Figure 3A). Epididymal weights were decreased by 18% at the highest dose of ENU, with smaller decreases at lower dose levels. These decreases in epididymal weight were not statistically significant.
WT CP
Wild-type mice exposed to 60 mg/kg/day CP had 19% and 18% decreases in paired testes and epididymal weights, respectively, relative to controls (Figure 3A). Effects on testis weights were statistically significant. Reproductive organ weights were not affected at lower doses of CP.
NULL ENU
Paired testis weights were significantly decreased by 55% at 60 mg/kg/day ENU, but were not altered at ≤20 mg/kg/day (Figure 3B). Similarly, epididymal weights were decreased by 23% at 60 mg/kg/day ENU, but were not affected at lower dose levels.
NULL CP
In NULL mice, CP treatment did not significantly alter testis or epididymal weights (Figure 3B).
Phase II: Testicular Histology
Photomicrographs illustrating testicular histopathology appear in Figure 4. Consistent with previous reports (Chandrasekaran and Richburg 2005), testicular histology was similar in WT and NULL control mice, with both genotypes exhibiting normal-appearing seminiferous tubules lined with Sertoli cells, spermatogonia, primary and secondary spermatocytes, and spermatids (e.g., histology in NULL controls shown in Figure 4A and 4B).
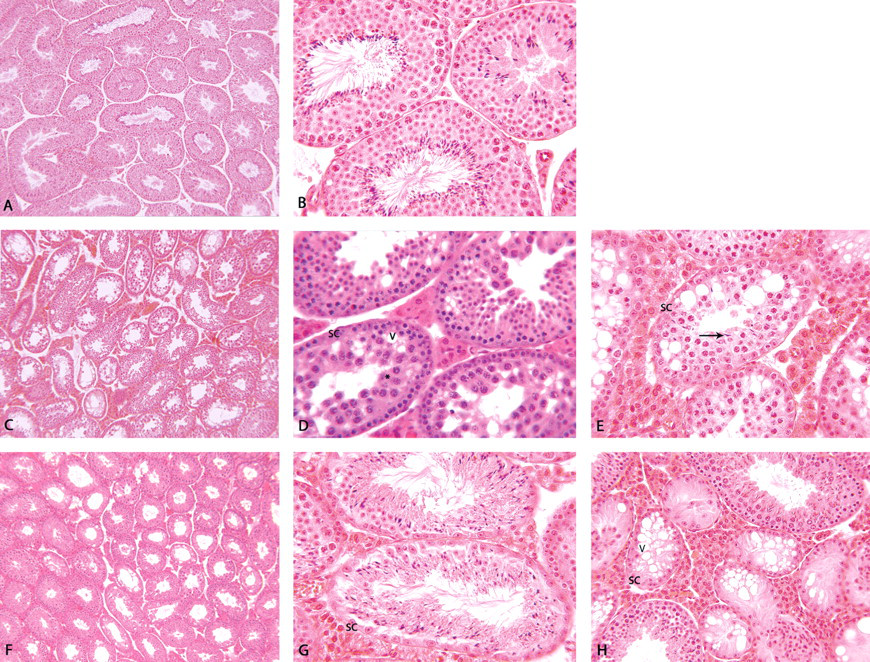
Testicular histology was similar in phase II control WT and NULL mice at 10× (A–NULL mice) and 40× (B–NULL mice). Seminiferous tubules from WT mice treated with 60 mg/kg/day ENU had severe degeneration with depletion of the elongating steps of spermatid development and vacuolization of Sertoli cells (C–10×, D–400×). Where spermatids remained, they often lacked proper radial orientation (E–400×). Seminiferous tubules from NULL mice given 60 mg/kg/day ENU also had severe degeneration, with the most common effect being tubules lined with late-stage or mature spermatids with only a rim of Sertoli cells (F–10×, G–400×). With CP treatment (60 mg/kg/day), there was a loss of germinal epithelium in approximately 20% of the seminiferous tubules in NULL mice, with some Sertoli cell vacuolization (H–250×). Similar effects were seen in WT mice at 60 mg/kg/day CP.
WT ENU
With ENU treatment at 20 or 60 mg/kg/day, WT mice exhibited moderate to severe degeneration in many of the seminiferous tubules, with complete depletion of the elongating steps of spermatid development. The germinal population of numerous seminiferous tubules consisted only of spermatogonia, pachytene, and leptotene/zygotene spermatocytes (Figure 4C and 4D). Some tubules had an additional layer of immature round spermatids lining the lumina. Mature spermatids were rarely present, and some of the mature spermatids lacked proper radial orientation (Figure 4E). Many tubules had prominent vacuolization of Sertoli cells, and occasionally, seminiferous tubules were lined only by Sertoli cells. In WT mice exposed to 60 mg/kg/day, the majority of seminiferous tubules were altered, whereas at 20 mg/kg/day, ~10–50% of tubules were affected.
WT CP
At 60 mg/kg/day CP, < 50% of WT mice showed altered testicular histology. In affected WT mice, there was a complete loss of germinal epithelium and some Sertoli cell vacuolization was present. Effects were similar to those seen in high-dose NULL mice (discussed below). Testicular histology was not affected in WT mice treated with 6 or 20 mg/kg/day CP.
NULL ENU
Ethylnitrosourea-treated NULL mice also had moderate to severe degeneration of the seminiferous tubules at 60 mg/kg/day; however, this degeneration was evidenced by loss of several phases in the maturation progression of spermatogonia. The most common effect was tubules lined with late-stage or mature spermatids with only a rim of Sertoli cells. These tubules were notable for their lack of late-stage spermatocytes and early spermatids (Figure 4F and 4G). Mature spermatids lost their radial orientation in severely affected seminiferous tubules. Sertoli cell vacuolization was present in a few tubules. At 60 mg/kg/day ENU, fewer seminiferous tubules were affected than in WT mice, and the damage was generally less severe. There were no effects on testicular histology in NULL mice at doses ≤20 mg/kg/day ENU. Thus, ENU affected different populations of spermatogenic cells depending on the zygosity of the treated mice.
NULL CP
Effects at 60 mg/kg/day CP were similar in WT and NULL mice, although the effects were slightly less in NULL mice. In the two affected CP-treated NULL mice, there were decreases in the numbers of germinal epithelial cells in approximately 20% of the seminiferous tubules (Figure 4H). The remaining tubules were within normal limits. Sertoli cell vacuolization was seen in these affected animals. Testicular histology was not affected in NULL mice treated with < 20 mg/kg/day CP.
Phase II: Epididymal Sperm Counts
WT CP and ENU
In WT mice, epididymal sperm counts (total sperm/g cauda) were decreased in a dose-dependent manner with both CP (20 and 60 mg/kg/day) and ENU (20 and 60 mg/kg/day) treatment (Table 1 ).
Phases I and II: Epididymal sperm concentrations
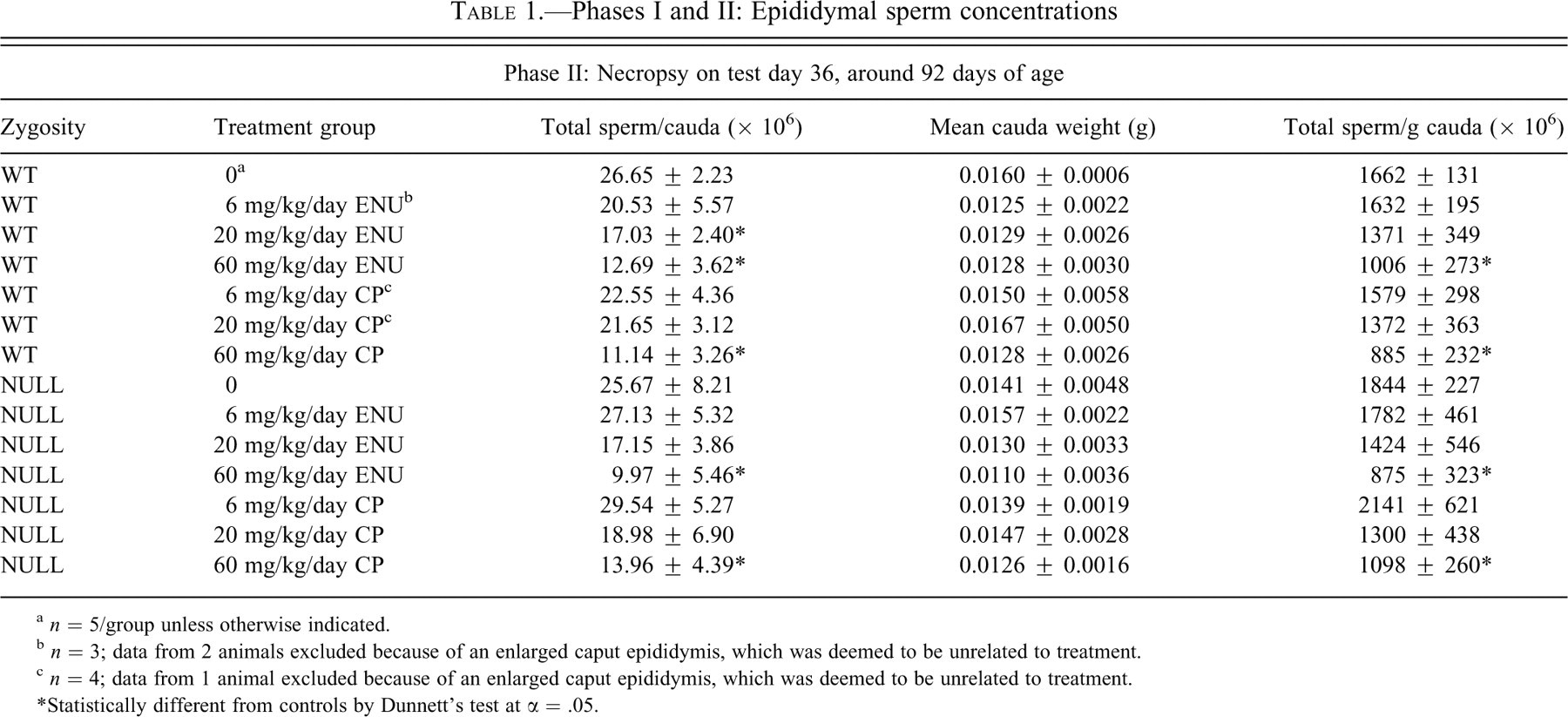
a n = 5/group unless otherwise indicated.
b n = 3; data from 2 animals excluded because of an enlarged caput epididymis, which was deemed to be unrelated to treatment.
c n = 4; data from 1 animal excluded because of an enlarged caput epididymis, which was deemed to be unrelated to treatment.
* Statistically different from controls by Dunnett’s test at α = .05.
NULL CP and ENU
In NULL mice, sperm counts were not altered at the lowest dose of either mutagen; however, counts were decreased at higher doses of ENU (20 and 60 mg/kg/day) and CP (20 and 60 mg/kg/day) (Table 1).
Phase II: Sperm Comet Assays and Apoptosis
To examine the influence of mutagen treatment and p53 zygosity on the integrity of sperm DNA, sperm obtained from the epididymides of control, ENU- and CP-treated WT and NULL mice on TD 36 were examined for the presence of DNA strand breaks using the comet assay and the percentage of apoptotic cells using the DNA diffusion assay.
WT ENU
Wild-type mice displayed a dose-related increase in DNA migration length with ENU treatment. At 6 mg/kg/day ENU, DNA strand breaks increased 37%, which was not statistically different from WT controls; however, DNA strand breaks were significantly increased by 61% and 85% at 20 and 60 mg/kg/day ENU, respectively (Figure 5A). Percentage of apoptosis was not significantly increased at any dose of ENU (Figure 5B).
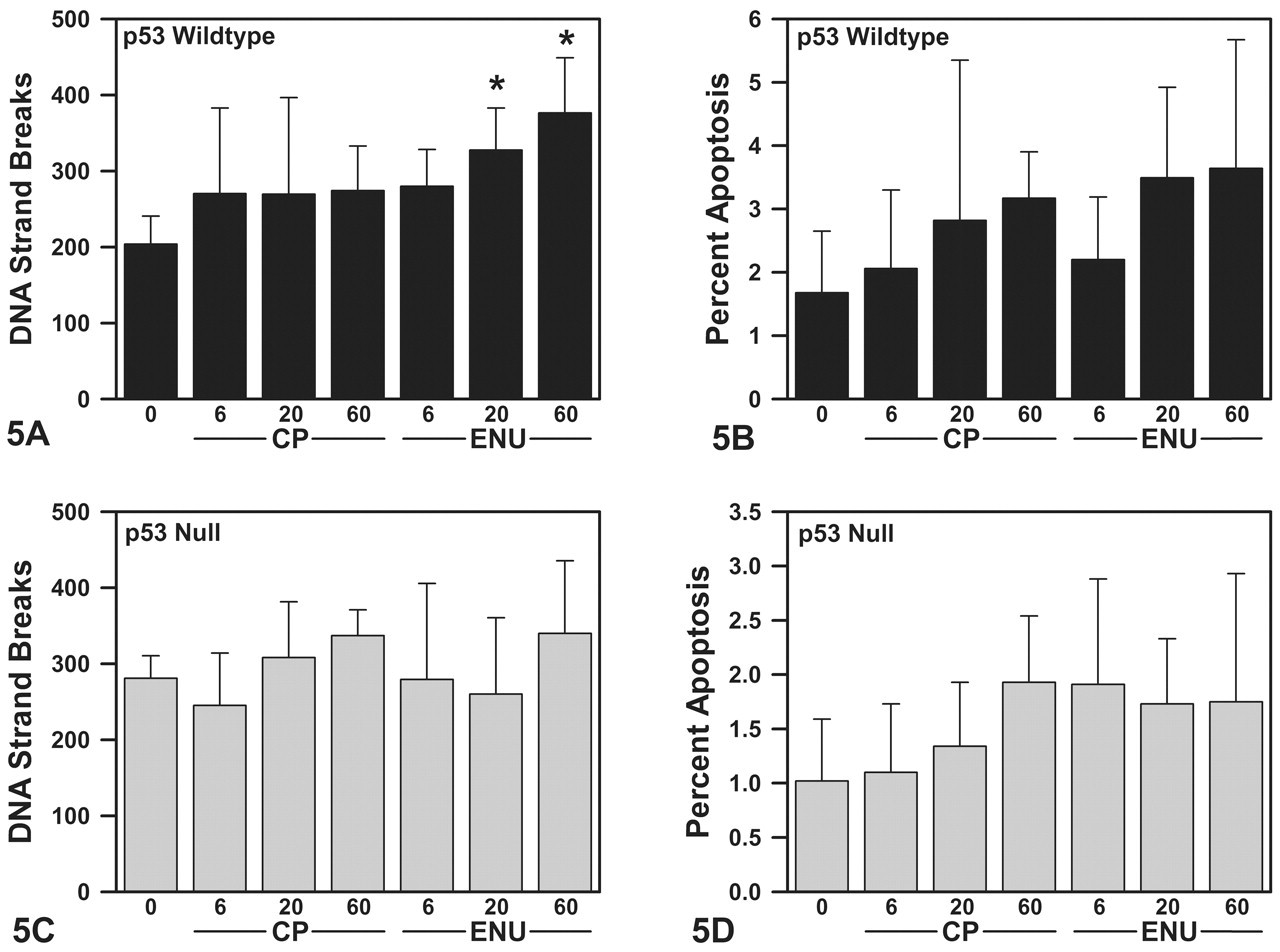
DNA strand breaks in the comet assay (A - WT, C - NULL) and apoptosis in the DNA diffusion assay (B: WT, D: NULL) in sperm from WT and NULL mice on TD 36. Ethylnitrosourea-treatment increased strand breaks at 20 and 60 mg/kg/day doses in WT mice (A); however, apoptosis was not statistically increased (B). There were no significant treatment-related changes in DNA strand breaks or apoptosis in response to ENU treatment in NULL mice or CP treatment in mice of either zygosity. Fifty cells were analyzed from each animal for average Comet Extent. One thousand cells from each animal were analyzed for percent apoptotic sperm. Data are expressed as means + SD. Asterisks indicate values that were significantly different from control by ANOVA and Dunnett’s test at α = .05 (n = five/group).
WT CP
In WT mice, CP treatment did not significantly increase the frequency of DNA strand breaks (Figure 5A), which was relatively consistent across all CP doses (32–35%). Furthermore, the percentage of apoptotic sperm was not significantly altered with CP treatment (Figure 5B).
NULL ENU
Ethylnitrosourea-treated NULL mice did not exhibit a significant dose-related effect on DNA strand breaks (Figure 5C), exhibiting 0.5% and 7% decreases in DNA strand breaks at 6 and ≤20 mg/kg/day ENU, respectively, and a 21% increase in strand breaks at 60 mg/kg/day ENU. Sperm apoptosis was not significantly increased at any dose of ENU (Figure 5D).
NULL CP
Similar to the WT mice, neither DNA strand breaks nor apoptosis were significantly increased at any dose of CP in NULL mice (Figure 5C and 5D). With 6 mg/kg/day CP, DNA strand breaks decreased 13%, but increased 10% and 20% at 20 and 60 mg/kg/day CP in NULL mice.
Discussion
This study was designed to examine the role of p53 in the repair of DNA damage induced in germ cells by ENU and CP. It was hypothesized that if p53 is critical for the repair of DNA damage in developing sperm or is involved in removing damaged sperm via apoptosis, then p53 NULL mice should exhibit greater germ cell damage than p53 WT mice following mutagen exposure.
Results from the current study are summarized in Table 2 . In phase I, epididymal sperm counts were not significantly altered on TD 4 in WT or NULL mice, indicating that the mutagen treatments were not acutely toxic to mature sperm. However, migration of sperm DNA was slightly increased in WT mice and decreased in NULL mice with high-dose ENU or CP treatment. The increased DNA migration demonstrated increased DNA strand breaks in WT mice, whereas the decreased DNA migration in NULL mice indicated fewer DNA strand breaks or DNA cross-linking. These data suggest that mutagen treatment, although not acutely cytotoxic, had effects on the DNA of mature epididymal sperm. Notably, results from these TD 4 samples do not address whether earlier stages in the spermatogenic cycle were affected by mutagen treatment, as the TD 4 sperm assessment would not allow sufficient time for these cells to mature. Testicular damage would remain undetected in the collected epididymal sperm used in the phase I analyses.
Results summary
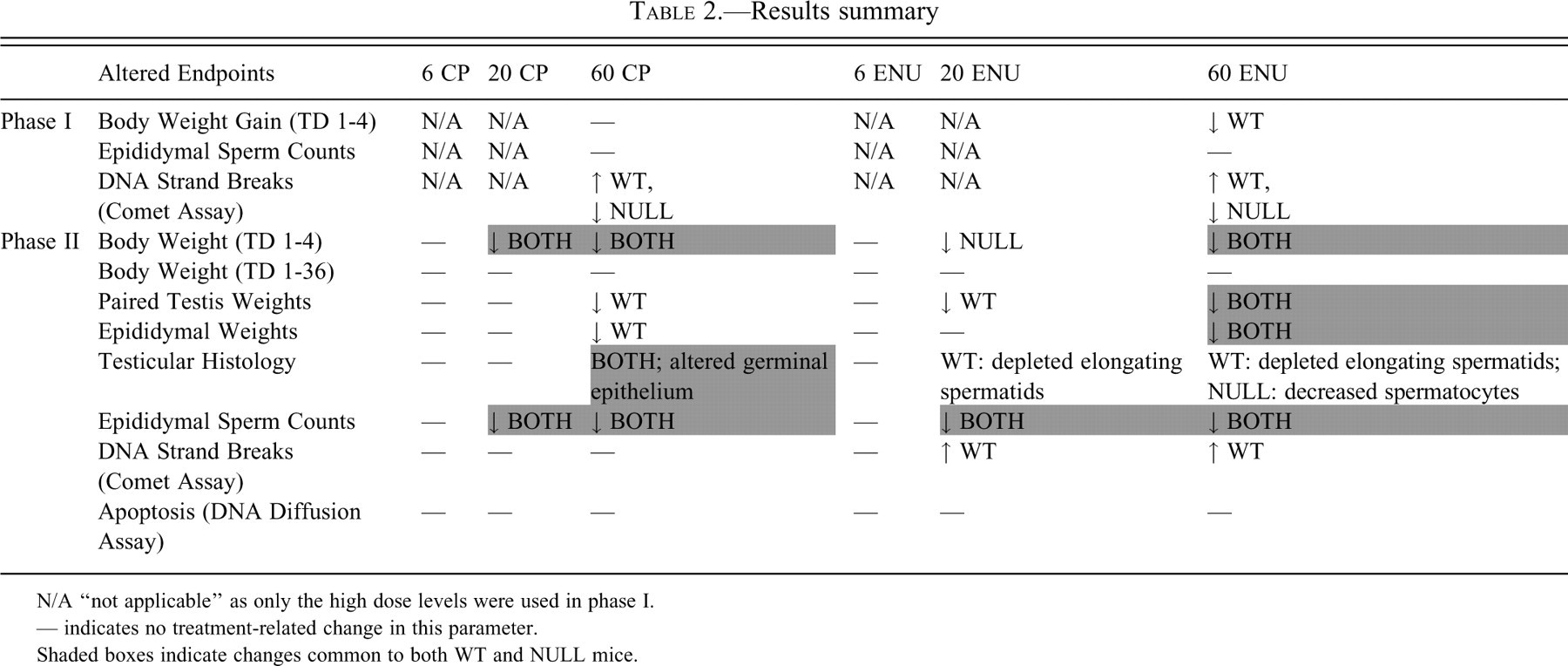
N/A “not applicable” as only the high dose levels were used in phase I.
--- indicates no treatment-related change in this parameter.
Shaded boxes indicate changes common to both WT and NULL mice.
In phase II, numerous end points (reproductive organ weights, testicular histology, sperm counts, sperm DNA strand breaks, and apoptosis) were examined thirty-six days after initiation of mutagen treatment to determine whether unrepaired DNA damage remained in WT and NULL mice, particularly in sperm that have matured from exposed spermatocytes. Our sampling scheme was specifically designed to detect damaged sperm cells that remained, despite having an opportunity for repair after mutagen exposure as spermatocytes. This portion of the spermatogenic cycle was targeted, because P53 is thought to play an active role in spermatocyte repair, as it is highly expressed in the pre-leptotene/leptotene/early pachytene stages (Bakalkin et al. 1994; Lee et al. 1995; Oberosler et al. 1993; Schwartz et al. 1993; Sjoblom and Lahdetie 1996). Thus, it was hypothesized that differences between WT and NULL mice would be most easily detected by focusing on mature cells from this stage. One assumption in our sampling scenario is that neither the spermatogonial cell cycles nor the rate of spermatogenesis were markedly altered by four-day treatment with CP or ENU. This assumption has been supported in other studies in which mice were exposed to chemical mutagens (Adler 1976; Oakberg et al. 1982), but it was not evaluated in the current study. Although there is evidence of a p53-dependent premeiotic delay in response to low levels of DNA damage (Schwartz et al. 1999), timing of the spermatogenic cycle is tightly controlled, allowing only minimal cell cycle arrest at any given stage (Sjöblom, West, and Lähdetie 1998).
In the present study, the results suggest that CP treatment produced responses that were primarily p53 independent. The pattern of responses to CP treatment was similar in both p53 WT and NULL mice (Table 2). Both genotypes had decreased epididymal sperm counts at 20 and 60 mg/kg/day, suggesting that p53-independent apoptotic pathways may have been operating at both dose levels. Testicular histology indicated that the germinal epithelium was altered in both genotypes. These data indicate that p53 status did not affect the murine response to CP treatment and that multiple apoptotic pathways in testicular germ cells may explain the inability of NULL mice to prevent apoptosis with CP treatment. This result is consistent with the findings of Sjöblom, West, and Lähdetie (1998), who did not detect any increase in testicular p53 or subsequent signaling molecules (p21, mdm) after exposing rats to three different chemical mutagens. In contrast to WT mice, reproductive organ weights were decreased to a lesser degree in NULL mice given 60 mg/kg/day CP, which may be the result of a difference in sensitivity and/or dynamic range in NULL mice.
In contrast to CP, data suggest that the response to ENU treatment may have involved a more primary role for p53. This finding is particularly evident at lower doses of ENU, where more effects were seen in WT mice than NULL mice in phase II of this study. For example, testis weights were significantly altered in WT mice at 20 mg/kg/day ENU, whereas this parameter was not affected in NULL mice until 60 mg/kg/day. This finding is consistent with data published by Schwartz et al. (1999), in which p53 was shown to be most important in the maintenance of sperm quality when low-level DNA damage was present. As the damage became more severe, it is conceivable that p53-independent pathways were activated, which would be consistent with the decreases in testis weight that was seen in both zygosities at 60 mg/kg/day ENU. Overlapping checkpoints and apoptotic pathways would be the most robust means to ensure maintenance of genomic integrity.
Testicular histology also was altered at lower doses of ENU in WT mice than NULL mice. In control animals, p53 NULL mice showed no differences in testicular histology relative to WT mice, consistent with the results reported by Chandrasekaran and Richburg (2005). However, at 20 and 60 mg/kg/day ENU, WT mice were noted to have seminiferous tubules with depleted elongating steps of spermatid development. In NULL mice, treatment-related changes in testicular histology were seen only at 60 mg/kg/day, and cells at different spermatogenic stages were affected. High-dose NULL mice had seminiferous tubules lined with late-stage spermatids and Sertoli cells, with a depletion of late-stage spermatocytes and early stage spermatids. These histopathology findings suggest that the long-term response of WT and NULL mice differed following ENU exposure.
In WT mice, with all quality control (QC) pathways intact, late-stage spermatids were missing on TD 36. This observation suggests that damaged spermatogonia were removed from the testes at some stage during this thirty-six-day interval. It has been reported that apoptosis of differentiating spermatogonia is p53 dependent (Hasegawa et al. 1998) and loss of spermatid/spermatozoa is consistent with 34.5-day period required for type A/intermediate spermatogonia to mature to spermatozoa (Oakberg 1956). Aside from the spermatid loss, the testes in WT mice showed resumption of the spermatogenic cycle by TD 36 with the presence of developing spermatogonia and spermatocytes; thus, primary type A spermatogonial stem cells were available to repopulate the testes. In this case, it is possible that WT mice would recover from ENU-induced lesions. In contrast, if NULL mice experienced similar damage to type A/intermediate spermatogonia as was seen in WT mice, these damaged spermatogonia were not removed from the testes, but rather developed into late-stage spermatids. This finding is consistent with the heat stress hypothesis, proposed by Ohta, Aizawa, and Nishimune (2003), that p53-dependent apoptosis is critical in differentiated spermatogonia and spermatocytes. Furthermore, NULL mice exhibited a loss of late-stage spermatocytes and early stage spermatids, cell stages that had recovered in WT mice. These data suggest that testes in NULL mice did not recover as efficiently as the testes in ENU-treated WT mice. The apparent depletion of primary type A spermatogonial stem cells makes recovery seem unlikely in ENU-treated NULL mice; thus, once the remaining elongated spermatids develop into mature sperm, the testes may progress to end-stage atrophy in these animals. Dampened recovery of the testes in NULL mice has been reported previously. Radiation experiments have shown decreased recovery of the germinal epithelium in the testes of NULL mice (van Buul et al. 2002), including the production of fewer differentiated progeny from stem spermatogonia (Hasegawa et al. 1998).
It is possible that ENU treatment damaged Sertoli cells in both WT and NULL mice, as Sertoli cell vacuolization was noted in affected tubules during histological examination. Damage to Sertoli cells may have resulted in secondary killing of spermatogenic cells (Parvinen 1982). Thus, Sertoli cell functional changes cannot be ruled out as a contributor to ENU testicular responses in WT and NULL mice.
With epididymal sperm, sperm counts were decreased in both WT and NULL mice at 20 and 60 mg/kg, despite differences in testicular histopathology across these groups. It is important to note that cells sampled in the cauda epididymis were approximately one week further along the maturational pathway at the time of exposure. Thus, cells exposed as type A spermatogonia became part of the testicular histology samples (elongated spermatids), whereas cells exposed as spermatocytes became part of the sperm sampled in the cauda epididymis for sperm counts, DNA strand breaks, and apoptosis. If both p53-dependent and -independent pathways were removing damaged spermatocytes in WT and NULL mice, then similar effects on sperm counts are possible. Aside from the targeted spermatocytes, it is clear that additional spermatogenic cell types were affected, resulting in different histopathological outcomes in NULL and WT mice.
DNA strand breaks were significantly increased in WT mice at 20 and 60 mg/kg/day ENU, whereas ENU did not significantly increase strand breaks in NULL mice. This finding differs from the report by Schwartz et al. (1999), who noted lower in vivo DNA repair and more damaged DNA in mature sperm in NULL mice after irradiation or cisplatin treatment. It is possible that the type of DNA damage differs between those stressors and ENU and that the type of DNA lesion contributes to different responses in these two cases. We speculate the presence of DNA crosslinks in sperm of ENU-treated NULL mice could have retarded electrophoretic mobility of DNA, leading to an underestimation of DNA strand breaks. Another possibility is that sperm cells with high DNA damage could have been eliminated from ENU-treated NULL animals by p53-independent pathways prior to reaching the epididymis for sperm collection.
Treatment-related alterations in apoptosis were not seen in either WT or NULL mice. The failure of ENU to significantly increase sperm apoptosis in WT and NULL mice is not inconsistent with the decrease in epididymal sperm counts seen at 20 and 60 mg/kg/day ENU. It is possible that testicular germ cells in ENU-treated mice underwent cell loss at an earlier spermatogenic stage in the testis prior to the sperm apoptosis assessment in the epididymis. Apoptosis at the spermatid stage is believed to be p53 independent (Ohta, Aizawa, and Nishimune 2003). Spermatids were present in the testicular histology samples on TD 36; however, it is difficult to determine whether spermatids were lost in an earlier generation of ENU-exposed spermatocytes. Alternatively, apoptosis was estimated in sperm samples that were transported overnight on ice in medium. Thus, reported incidence of apoptosis may be an underestimation owing to loss of apoptotic cells. Seaman et al. (2003) has shown long-term elevated levels of cisplatin-induced germ cell apoptosis in male mice. The lower dose of cisplatin produced more germ cell apoptosis than did the higher dose because of cell loss. For NULL mice, the lack of an increase in apoptosis is consistent with a previous report showing that the absence of p53 prevents germ cells from undergoing apoptosis following irradiation (Hasegawa et al. 1998).
Together, these differences in the responses of WT and NULL mice suggest that WT mice may be mounting a p53-mediated response to ENU-induced germ cell damage. The correlative increase in sperm strand breaks in WT mice may indicate that p53-mediated pathways are contributing to a process to remove sperm with extensive DNA damage. NULL mice did not have increased strand breaks in epididymal sperm. This finding may indicate (1) less damage in epididymal sperm from NULL mice (i.e., less sensitive to ENU treatment), a possibility not supported by the epididymal sperm count data; (2) less damage in epididymal sperm from NULL mice owing to removal of damaged sperm at an earlier stage of spermatogenesis, a possibility with some support from the epididymal sperm count data; or (3) similar damage in the sperm from NULL mice, but the DNA damage is of a different type and therefore, not detected as strand breaks. Without extensive damage, these sperm may not be subject to apoptosis. If this were the case, sperm with damaged DNA would be more likely to survive in NULL mice. This hypothesis has been proposed previously (Kunugita et al. 2002; Schwartz et al. 1999) and is consistent with the decreased reproductive efficiency seen in NULL mice. It is possible that multiple factors contributed to the observed results (e.g., perhaps some damaged sperm were removed earlier in the spermatogenic cycle, but remaining sperm contain DNA with greater damage). This idea would suggest that p53-independent pathways facilitate maintenance of germ cell integrity, but that these pathways are not as efficient as a full complement of repair/apoptosis pathways that include p53.
Within the testes, there are believed to be multiple checkpoints that operate at different stages in germ cell maturation and trigger germ cell death in an effort to maintain germ cell integrity. These checkpoints operate in spermatogonia (Allan, Gobe, and Harmon 1987, 1992; Huckins 1978) and in spermatocytes and spermatids (Blanco-Rodriguez and Martinez-Garcia 1996; Kerr 1992). Within these checkpoints, both p53-dependent and p53-independent pathways have been identified. In response to detectable levels of germ cell damage at a given checkpoint, apoptosis is initiated to remove the damaged cells.
The data in the current study indicated a greater response to ENU treatment in WT mice, which are able to mount a p53-mediated response to initial, low-level damage. However, when greater damage is induced, redundant apoptotic (p53-independent) pathways are operational in the maintenance of germ cell genomic integrity. This finding is consistent with the cell loss in the testes and epididymides of NULL mice treated with 60 mg/kg/day ENU. The conditions under which p53 is critical for successful spermatogenesis are disputed. Some investigators have postulated that p53 in critical for full apoptotic potential to be achieved in the developing germ cells. Some evidence exists to suggest that p53 function is not fully substitutable in spermatogenesis (Rotter et al. 1993). Other researchers have postulated that p53 is important for efficient apoptosis. In these cases—for example, mono-(2-ethylhexyl)phthalate-induced testicular damage (Chandrasekaran and Richburg 2005)—loss of p53 has been associated with less efficient DNA repair or apoptosis via p53-independent pathways. In the case of heat stress–induced testicular apoptosis (i.e., cryptorchidism), both p53-dependent and independent pathways are operational; however, apoptosis is observed three days later in NULL than WT mice (Yin et al. 2002). Other researchers have ascribed that the type of genetic damage dictates the necessity for p53-driven apoptosis. Certain types of stress (e.g., clastogenic damage) require p53 for effective apoptosis (Sansom and Clark 2000). Thus, it is possible that p53 plays different roles in DNA repair, depending on the cell stage that is affected (Hasegawa et al. 1998), the type of damage present (Tang, Willers, and Powell 1999), and the possible repair pathways available (Yin et al. 2002).
Thus, this study suggests that p53-dependent and -independent pathways are operational in the murine testes following exposure to mutagens. In the case of CP, p53-independent responses were more apparent, because testicular responses were similar in mice regardless of zygosity. In contrast, the presence of p53 influenced testicular response with ENU treatment. In WT mice treated with ENU, increased DNA strand breaks and different testicular histopathology were seen compared with ENU-treated NULL mice. These data suggest that WT mice mounted a p53-mediated response to ENU treatment. Still, compensatory pathways appear to be operational in NULL mice to remove damaged sperm, as shown by decreased epididymal sperm counts in both CP-and ENU-treated mice.
Footnotes
Acknowledgments
The authors gratefully acknowledge the assistance of Dr. Jon Hotchkiss in the preparation of this manuscript.