
Abstract
Rhabdomyosarcoma (RMS), a rare pediatric soft tissue sarcoma, seldom involves the external ear. Congenital ichthyosis vulgaris (IV), a genetic disorder causing hyperkeratosis, has not been previously linked to RMS. We report the first case of embryonal RMS with unique extension in a patient with IV. A 13 year-old male with ichthyosis presented with a 1 year history of a progressive lesion in the right intertragic notch, unresponsive to antibiotics and topical therapies. Imaging revealed a mass infiltrating the parotid gland, external auditory canal, and sternocleidomastoid muscle. Wide surgical excision with facial nerve preservation was performed. Histopathology confirmed embryonal RMS. Postoperative chemoradiotherapy was initiated, and metastatic workup showed no dissemination. This case highlights diagnostic challenges in rare RMS locations, particularly when masked by dermatoses like ichthyosis. The association between ichthyosis and RMS remains speculative; potential mechanisms include ichthyosis. Clinicians should maintain a high index of suspicion for malignancy in persistent cutaneous lesions.
Introduction
Rhabdomyosarcoma (RMS), a prevalent soft tissue sarcoma primarily affecting children, constitutes ~5% of all pediatric malignant neoplasms, 1 with an incidence of 4.8/million in this age group. 2 While RMS can arise in diverse anatomical regions, 35% to 40% of cases occur in the head and neck. 3 Histopathological classification includes embryonal (most common), alveolar, pleomorphic, and mixed subtypes, with alveolar RMS carrying the poorest prognosis. 4 RMS exhibits aggressive biological behavior, frequently leading to local invasion and distant metastasis. 5 The lungs are the most common site of distant spread, followed by the skeletal system, liver, and brain. 6 Treatment strategies—guided by tumor location, extent, and patient status—typically involve multimodal therapy with surgery, chemotherapy, and radiotherapy. 7 Early diagnosis is critical for improving outcomes 7 ; however, tumors in rare locations such as the outer ear pose diagnostic challenges. These lesions may mimic infectious or inflammatory processes, delaying appropriate management. 8 Notably, middle ear involvement is more frequently reported than external ear involvement in RMS. 9 Ichthyosis vulgaris (IV), a rare hyperkeratotic genodermatosis, is an inherited form of non-syndromic ichthyosis, with a very early age of onset. It is classified by clinical distribution, inheritance patterns, syndromal associations, and biochemical defects. 10 To our best knowledge, this represents the first reported association between congenital IV and RMS, with the latter demonstrating a unique extension pattern involving the external ear, parotid gland, and sternocleidomastoid muscle.
Case Presentation
A 13 year-old boy with congenital IV presented with a 1 year history of a wart-like lesion in the intertragic notch of the right pinna. The lesion had been treated with multiple courses of antibiotics and topical therapies without improvement and gradually enlarged over 11 months before referral to our ENT department. He denied otorrhea, otalgia, hearing loss, tinnitus, vertigo, facial weakness, dysphagia, odynophagia, fever, or weight loss. Physical examination revealed IV on the right ear, along with a soft, nodular, violaceous mass involving the right cavum concha and external auditory canal, extending to the retroauricular region as a 4 × 4 cm swelling. A 2 × 2 cm non-tender neck mass was also noted in the upper right cervical region (Figure 1). Contrast-enhanced Computed tomography (CT) of the head and neck demonstrated a lobulated soft tissue mass originating from the right auricle and external acoustic meatus, infiltrating the parotid gland and upper sternocleidomastoid muscle. The mass abutted the mastoid process without bony destruction but caused erosion of the auricle and external auditory canal cartilage. There was no middle or inner ear involvement or significant bony erosion (Figure 2a and b). Laboratory findings were unremarkable.
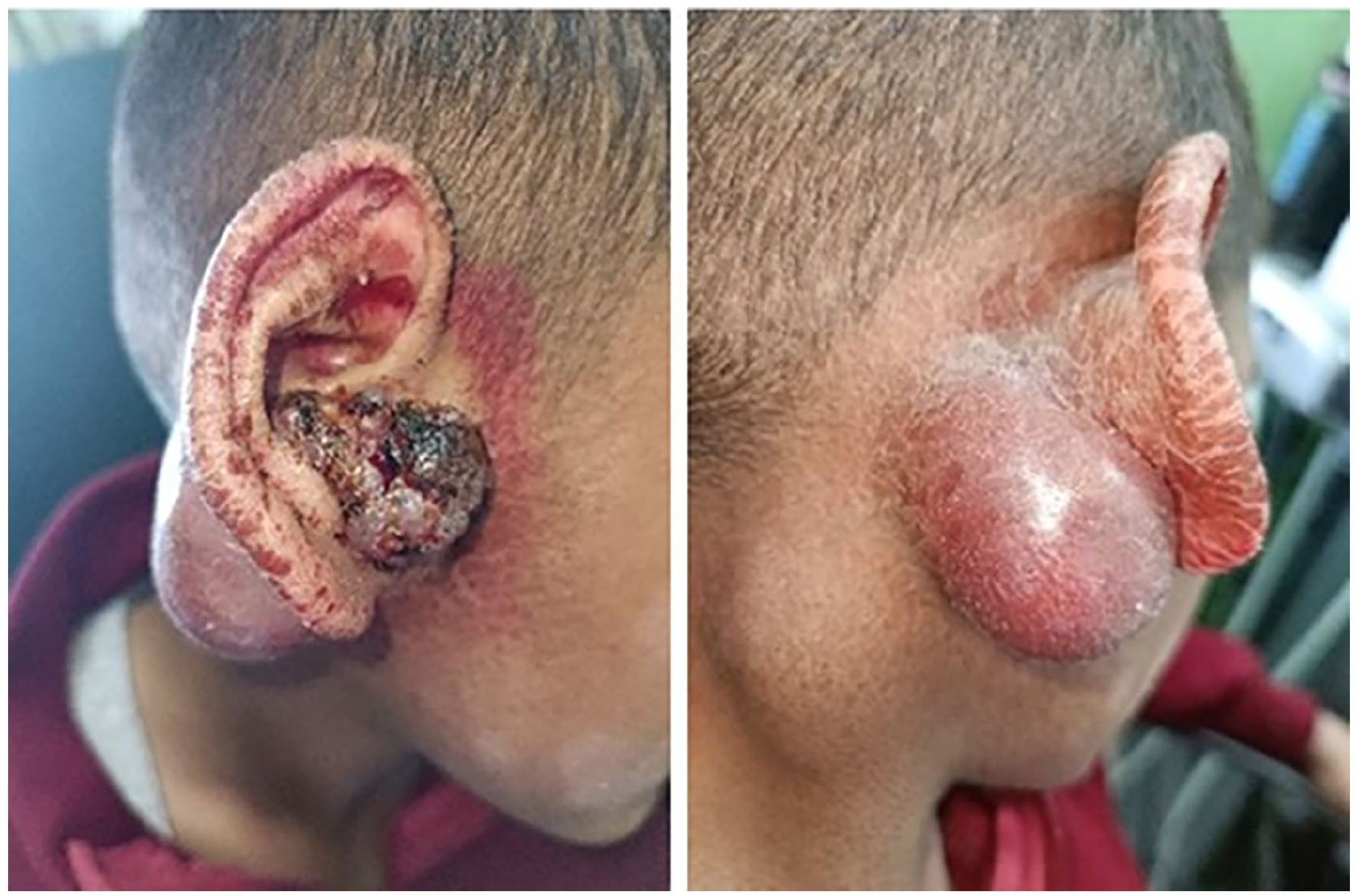
Demonstrating the extensive tumor mass involving the right auricle and external auditory canal, alongside associated ichthyosis vulgaris on the right ear.
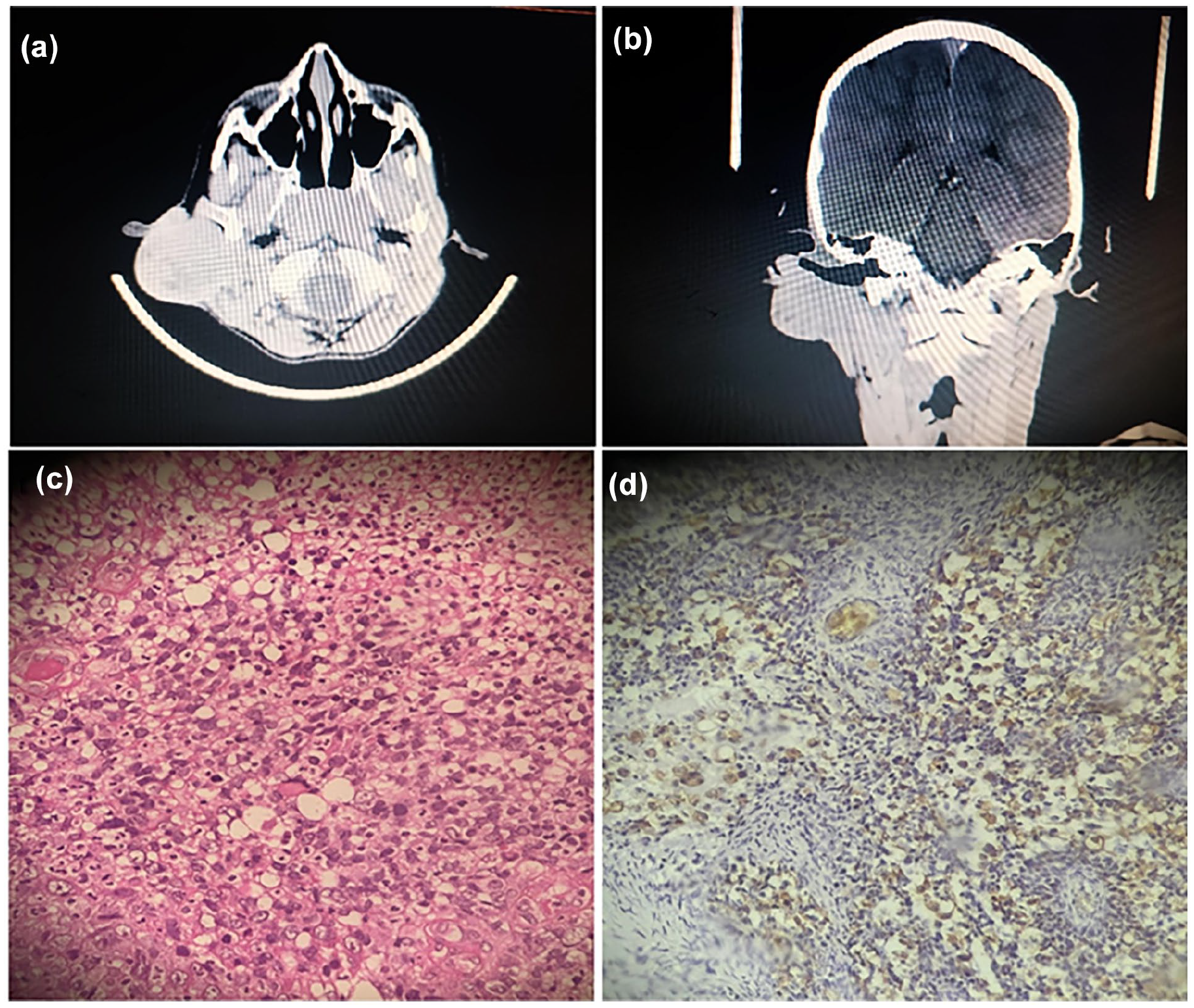
Contrast-enhanced CT scan of the head and neck demonstrates a lobulated soft tissue mass originating from the right auricle and external auditory canal, with infiltration into the parotid gland and upper sternocleidomastoid muscle (a, b). Sheets of spindle, round, and rhomboid cells with scant, deeply-eosinophilic cytoplasm, and anaplastic features (high nuclear-to-cytoplasmic ratio, hyperchromasia) (c), desmin positive staining (d).
Wide surgical excision under general anesthesia was performed. Intraoperatively, 1 cm safety margins were established around the tumor. The marginal branch of the facial nerve was identified near the main trunk, while the traditional retrograde identification approach was avoided due to the complete encasement of the main trunk by the tumor (Figure 3a). Total parotidectomy with facial nerve preservation and intraoperative stimulation was achieved. The tumor, infiltrated sternocleidomastoid muscle, and external auditory canal were excised en bloc (Figure 3b). Skin grafts from the neck and cheek were used for reconstruction (Figure 3c). Histopathology revealed sheets of round and rhabdoid cells with eosinophilic to clear cytoplasm, vesicular chromatin, pleomorphism, and focal anaplastic features. Immunohistochemical staining was positive for desmin (Figure 2c and d). A definitive diagnosis of embryonal RMS was made. Chemotherapy was initiated 2 weeks postoperatively. Follow-up head MRI 3 weeks post-resection showed no residual infiltration. Chest MRI and PET scan revealed no regional or distant metastases.
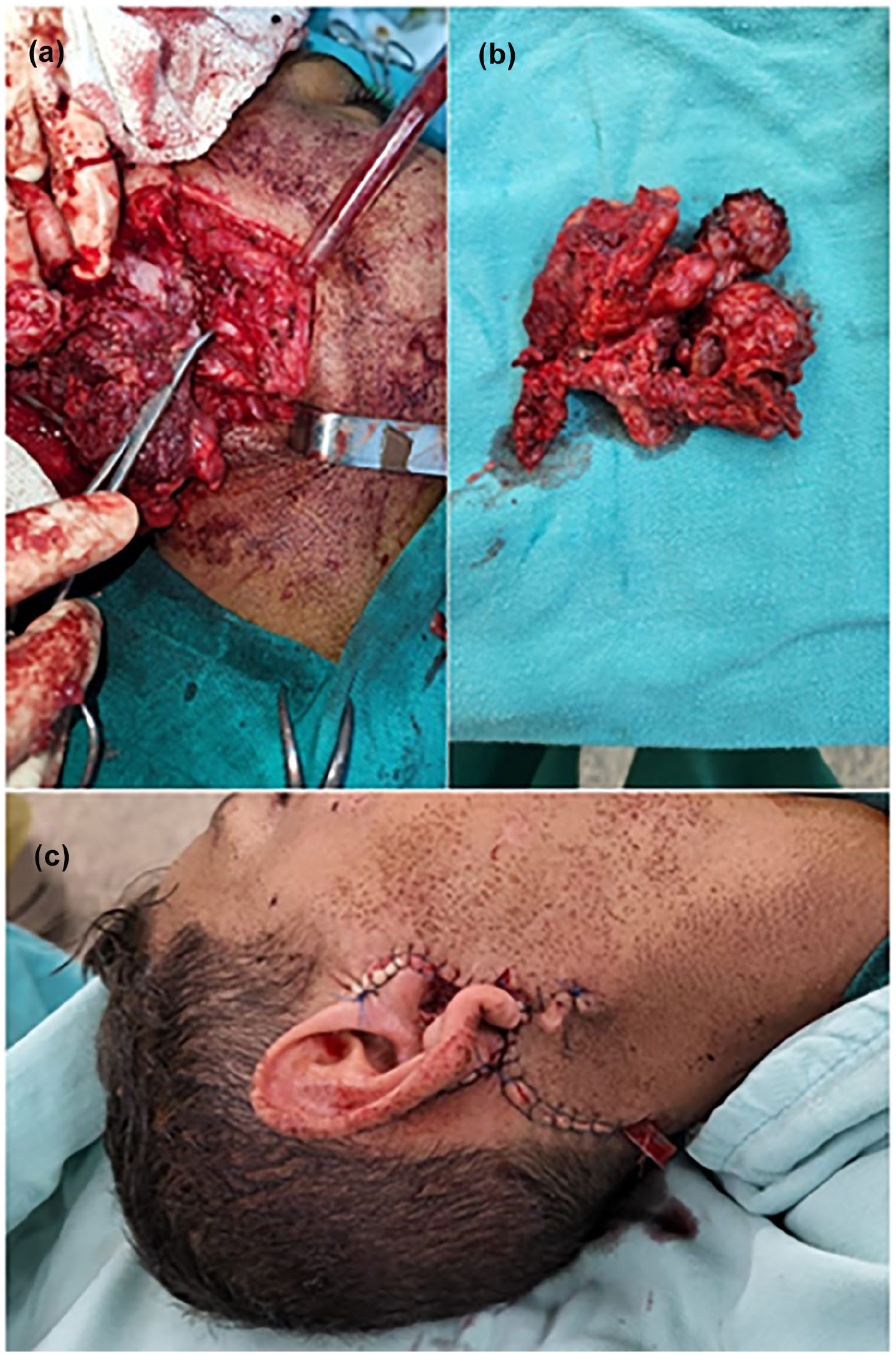
Intraoperative and postoperative views of the surgical management. Total tumor resection with parotid gland dissection, demonstrating preservation of the facial nerve (a, b). Postoperative reconstruction using skin grafts harvested from the neck and cheek (c).
Discussion
RMS, a rare soft tissue sarcoma originating from primitive mesenchymal cells, is the most common malignancy in the head and neck region among children, termed head and neck RMS (HNRMS). 1 HNRMS most frequently involves the orbits, oral cavity, pharynx, or neck, while external ear involvement is exceptionally rare.2,3 RMS is classified histologically (eg, embryonal, alveolar) and anatomically into orbital, parameningeal (involving regions adjacent to the meninges such as the middle ear, nasal cavity, or paranasal sinuses), and non-orbital/non-parameningeal subtypes. 4
This case, involving a 13 year-old boy with IV, was classified as non-orbital and non-parameningeal. The diagnosis of RMS is often delayed due to nonspecific symptoms mimicking common conditions.5,6 In this case, the lesion resembled a treatment-resistant skin wart. Clinical suspicion arose when the lesion grew rapidly despite antibiotics and topical therapies. Contrast-enhanced CT revealed a lobulated mass in the right auricle infiltrating the parotid gland and sternocleidomastoid muscle. Histopathological analysis of an excisional biopsy with 1 cm margins confirmed embryonal RMS, supported by positive immunohistochemical staining for desmin. Surgical management included total parotidectomy with facial nerve preservation, partial sternocleidomastoid resection, and reconstruction using neck or cheek skin grafts. Postoperative metastatic evaluation (brain/lung MRI, PET scan) showed no dissemination. Adjuvant chemoradiotherapy was initiated due to presumed microscopic metastasis and fragmented surgical margins.
This case represents the first reported association between congenital IV and RMS. While ichthyosis, a genetic disorder causing hyperkeratosis, is linked to keratinization-related skin tumors, its connection to non-cutaneous malignancies like RMS remains speculative. Proposed mechanisms include immunodeficiency or environmental factors (eg, arsenic exposure), though coincidental occurrence cannot be excluded. Vigilance for malignancies in ichthyosis patients with atypical lesions is crucial, warranting further research into genetic or environmental interactions.
Conclusion
This case represents the first documented association between congenital IV and RMS, highlighting the diagnostic and therapeutic challenges posed by rare tumor locations such as the external ear. The patient’s lesion, initially mimicking a benign wart, underscores the importance of maintaining a high index of suspicion for malignancy in persistent or atypical cutaneous growths, particularly in individuals with underlying genetic dermatoses. Multidisciplinary management—combining aggressive surgical resection, adjuvant chemoradiotherapy, and meticulous metastatic surveillance—was critical in achieving favorable outcomes. While the link between ichthyosis and RMS remains speculative, potential mechanisms such as immunodeficiency or environmental triggers warrant further investigation. This case emphasizes the need for heightened clinical vigilance in ichthyosis patients and advocates for research into genetic or environmental factors that may predispose to non-cutaneous malignancies. Such insights could refine diagnostic protocols and therapeutic strategies for rare tumor associations in the future.
Footnotes
Acknowledgements
The authors acknowledge the patient and his family for their cooperation.
Ethical Considerations
Written informed consent was obtained from the parents of the child for the publication of this case report.
Consent for Publication
Written informed consent was obtained from the parents of the child for the publication of this case report, including anonymized clinical details and any accompanying data.
Author Contributions
Mohammad Hamdi: conceptualized the case report, managed patient care. Yasser ALGhabra: collected clinical data, drafted the manuscript, and incorporated revisions. Kinana Jamal Hammoud: conducted literature review and analyzed diagnostic data. Mohammad Tanani: contributed to writing the discussion sections. Yahia Hamdi: collected clinical data. Arige Alassaf: supervised the clinical management, critically revised the manuscript for intellectual content, and approved the final version. All authors reviewed and approved the manuscript prior to submission.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the author or corresponding author upon reasonable request.