
Abstract
The main objective of this report is to describe a unique clinical case of a congenital cholesteatoma with complete lateral displacement of the cochlea and aggressive erosion of the internal auditory canal and lateral semicircular canals in a very young child. This report involves a 10-month-old female who initially presented with left intermittently bloody otorrhea at 2.5 months of age and audiology testing in the clinic demonstrated sensorineural hearing loss in the left ear. Initial computerized tomography (CT) revealed a destructive left temporal bone lesion displacing the left otic capsule and vestibular aqueduct with erosion and involvement of the inner ear structures including erosion into the cochlea and semicircular canals, and posterior fossa dura. A left transtemporal approach for cholesteatoma removal and ear canal closure was done. 4 months postoperatively, the patient required revision of the left transtemporal approach to the posterior fossa for recurrent cholesteatoma in the hypotympanum adjacent to the carotid artery and eustachian tube. This case is unique in the complete lateral displacement of the cochlea and aggressive erosion of the internal auditory canal and lateral semicircular canals in a very young child. It serves to remind otolaryngologists of the silent yet infiltrative nature of congenital cholesteatoma. The very young age of the patient and the presence of otorrhea at 1 month of age raise the question of whether this lesion could have been developing in utero. It lends support to the theory of developmental epithelial rests in congenital cholesteatoma.
Introduction
Congenital cholesteatomas are a rare, non-cancerous growth of squamous epithelium within the middle ear, most commonly diagnosed in children between the ages of 5 and 10 years old, although it can manifest at any age. In contrast to the more common acquired cholesteatomas which often occur secondary to repeated ear infections or Eustachian tube dysfunction, congenital cholesteatomas are thought to arise from epithelial remnants that fail to regress during fetal development, which can be of particular concern due to their ability to grow silently, often asymptomatic until significant damage to the middle ear structures occur. 1 Alternative theories of congenital cholesteatoma pathogenesis include the trapping of ectopic epidermal tissue during embryogenesis or the migration of squamous epithelium through a developmental anomaly in the tympanic membrane or Eustachian tube, although the exact pathophysiology is still an area of ongoing research. 2 The diagnosis of a congenital cholesteatoma can often be very challenging given the insidious onset and is often incidentally discovered during routine pediatric examination revealing subtle signs such as a retracted eardrum or a pearly mass behind the tympanic membrane. 3 Nonspecific presenting symptoms can include hearing loss, tinnitus, or otorrhea. Definitive diagnosis is often dependent upon imaging including high-resolution computed tomography (CT) and magnetic resonance imaging (MRI) which can provide detailed visualization of the mass in addition to its impact on surrounding structures. While CT scans are useful in assessing the bony involvement, MRIs are particularly applicable in differentiating the cholesteatoma from soft-tissue masses. The treatment of congenital cholesteatoma is typically with surgical intervention directed at removing the cholesteatoma, preserving hearing, and preventing later recurrence. The ideal surgical approach is often dependent upon the size, location, and extent of the cholesteatoma and can include procedures such as tympanomastoidectomy and either canal wall-up or canal wall-down procedures. 4 In cases with extensive involvement, hearing restoration procedures such as ossicular chain reconstruction can be done as well. Postoperative follow-up is a crucial aspect of congenital cholesteatoma management as well with studies showing recurrence rates of 10%-15% even after a seemingly successful initial surgery. 5 This case report explores a unique instance of congenital cholesteatoma in a young child, detailing the clinical presentation, diagnostic workup, surgical management, and postoperative outcomes. The necessary written informed consent was obtained from the patient’s legally authorized representative for the publication of this case report.
Case Report Description
A 10-month-old female presented with left intermittently bloody otorrhea at 2.5 months of age. The patient had a past medical history of recurrent otitis media with a history of pressure equalization tube (PET) placement in her right ear and failed PET placement in her left ear due to an obstructive lesion seen under sedation at the age of 5 months old. At presentation, she had been on multiple oral and topical otic antibiotics without resolution of otorrhea. On examination, her external auditory canals were impacted with bloody debris. Audiology testing in the clinic demonstrated sensorineural hearing loss in the left ear. Computerized tomography (CT) revealed a destructive left temporal bone lesion displacing the left otic capsule and vestibular aqueduct with erosion and involvement of the inner ear structures including erosion into the cochlea and semicircular canals, and posterior fossa dura (Figure 1A-C). The displaced cochlea in the external auditory canal is seen clearly specifically in Figure 1C.
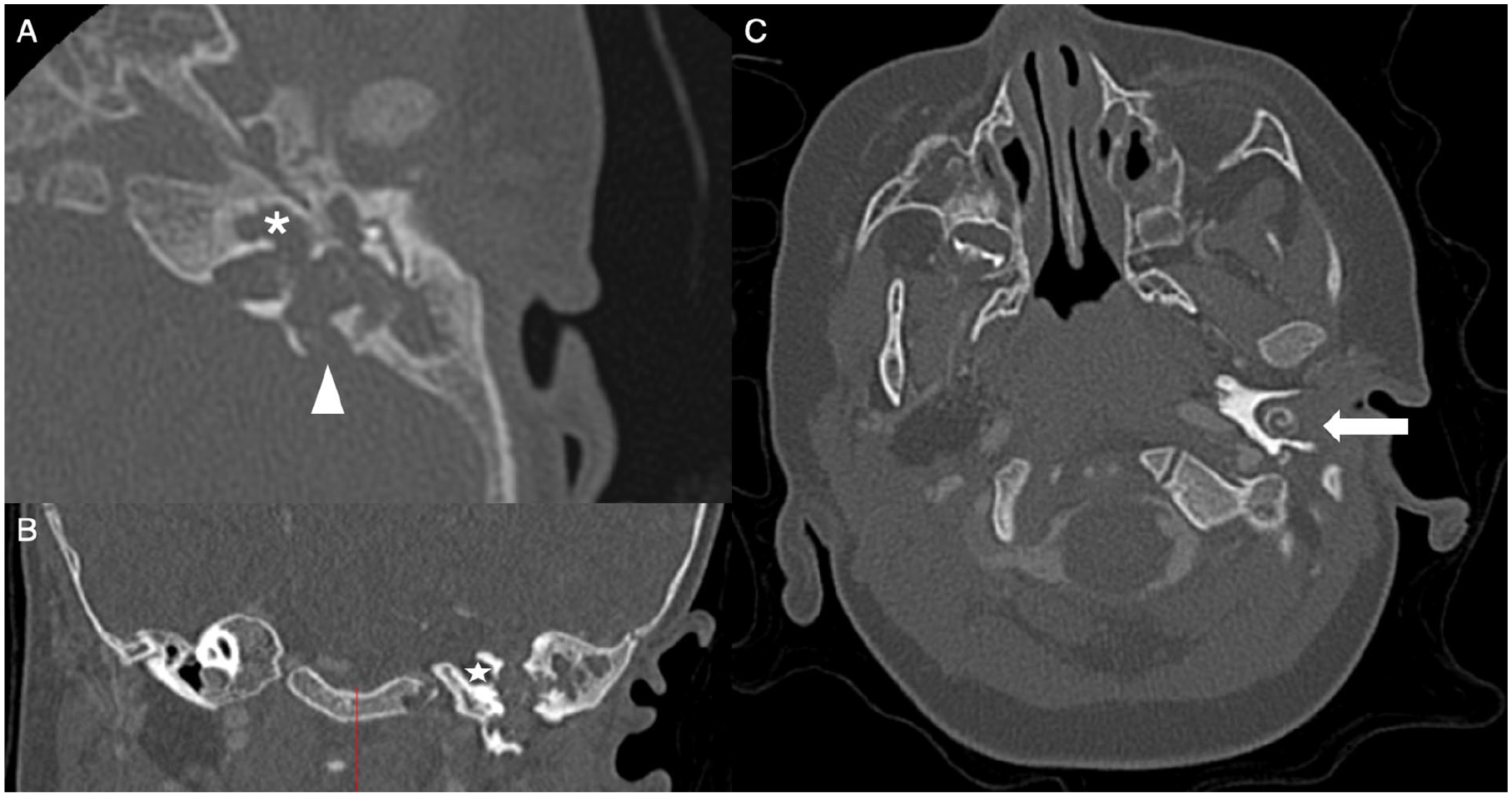
Preoperative CT (A) axial imaging of left middle ear and mastoid opacification with bony erosion of cochlea (asterisk), semicircular canals, and posterior dura (arrowhead); (B) coronal imaging lateral to the internal auditory canal (star) there is no cochlea; (C) the cochlea is displaced in the external auditory canal (arrow). CT, computerized tomography.
Treatment Description
At 10 months old, the patient underwent the left transtemporal approach for cholesteatoma removal and ear canal closure. There was a soft tissue tumor in the ear canal, mastoid, and middle ear space. The cochlea was found to be eroded and displaced laterally to the tympanic membrane annulus (Figure 2). Mastoidectomy revealed cholesteatoma and purulence against the dehiscent posterior fossa dura and along the dehiscent tympanic and mastoid segment of the facial nerve. Due to concerns over a potential cerebrospinal fluid (CSF) leak, the ear canal was closed. Surgical pathology confirmed the diagnosis of cholesteatoma, and the culture was positive for Pseudomonas aeruginosa and Escherichia coli. Given the close proximity of purulence against exposed dura, pediatric infectious disease was consulted and recommended 6 weeks of IV ceftazidime.
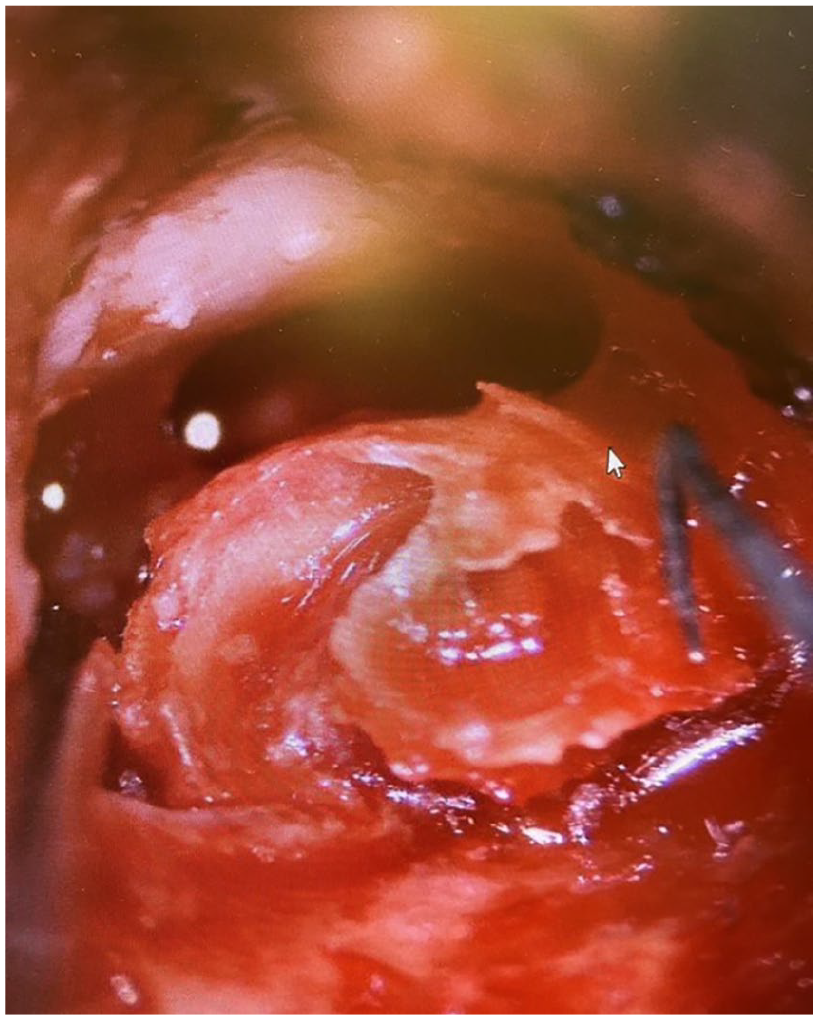
Intraoperative image of the eroded cochlea in the external auditory canal.
A follow-up MRI at 4 months after surgery showed focal 3-4 mm cholesteatoma in the mastoid tip. The patient was then taken back to the operating room for revision of the left transtemporal approach to the posterior fossa for residual cholesteatoma. Intraoperatively, there was cholesteatoma and purulence in the hypotympanum adjacent to the carotid artery (Figure 3A-D). Culture showed pan-sensitive pseudomonas which was treated with ciprofloxacin orally course for 14 days. The patient did well for a while until she developed left otalgia and scant otorrhea in the closed ear canal. She returned to the operating room at age 3, there was recurrent cholesteatoma in the hypotympanum adjacent to the carotid artery and eustachian tube, this was peeled off carefully. The mastoid tip was removed, allowing the removal of granulation tissue and cholesteatoma medial to the digastric muscle.
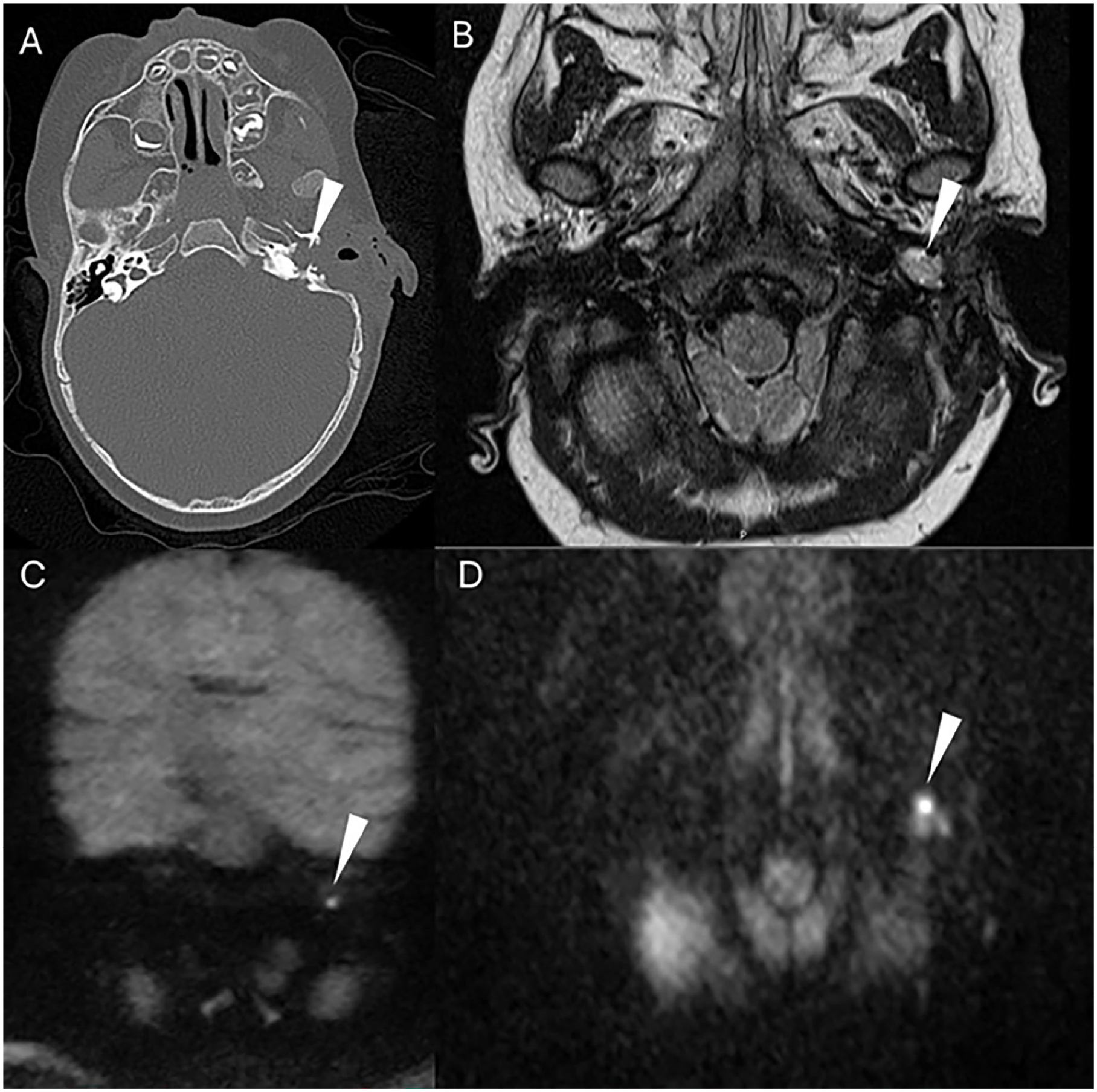
Imaging at 4 months after first surgery. (A) CT showed opacification adjacent to the carotid artery and eustachian tube, (B) MRI hyperintensity at the hypotympanic location, (C) and (D) MRI DWI sequence confirmed 3-4 mm residual cholesteatoma. MRI, magnetic resonance imaging.
Discussion
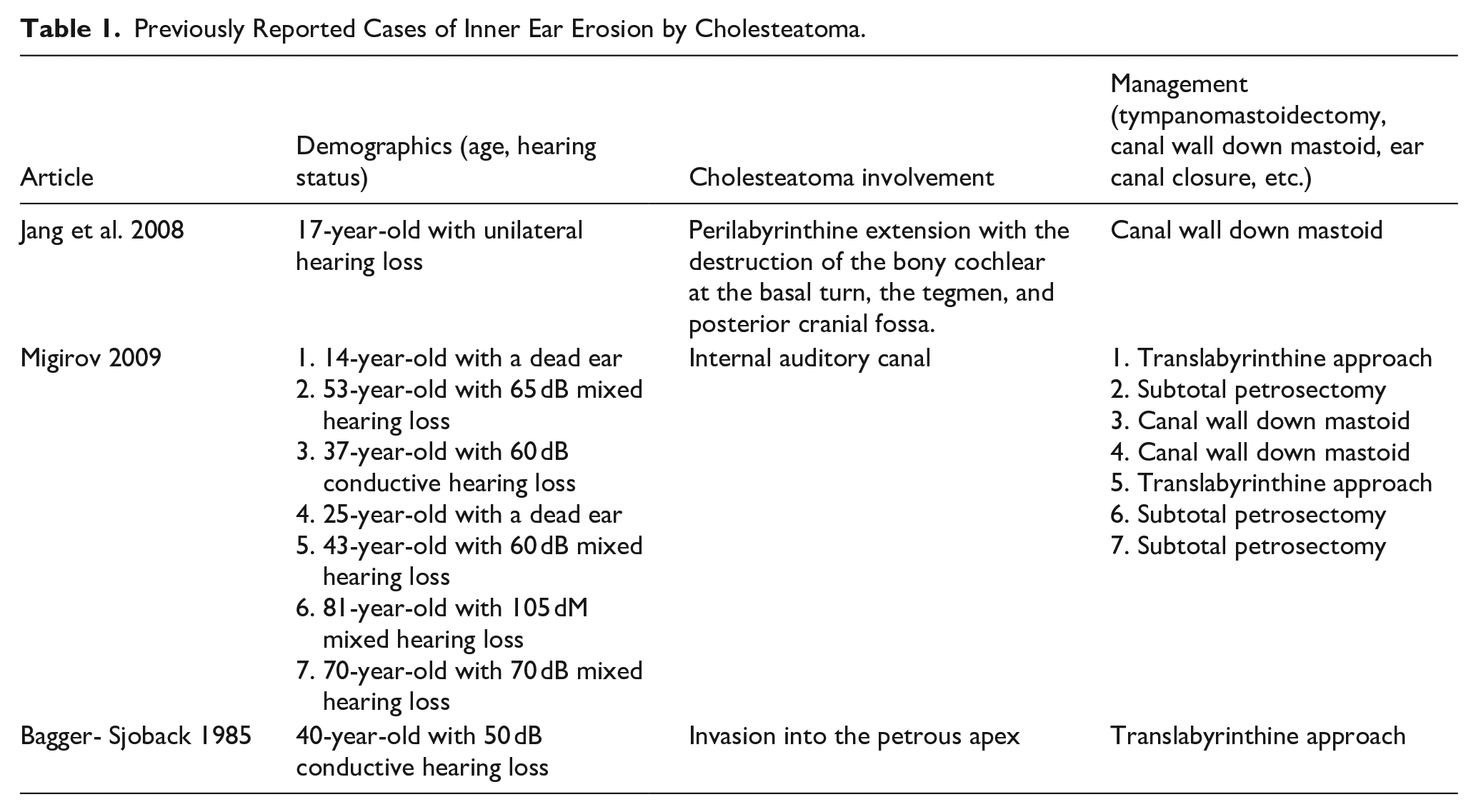
Congenital cholesteatomas (CC) of the temporal bone are thought to arise from squamous epithelial “rests.” 6 It is an uncommon disease entity, accounting for 4%-24% of all cholesteatomas with the average age at presentation being 4.9 years of age.7,8 Table 1 summarizes previously reported cases of inner ear erosion by cholesteatoma (Table 1). Specifically, CCs with extensive petrosal bone erosion are exceedingly unusual and are often asymptomatic.9,10 Common presenting symptoms for CC include hearing loss and otorrhea, although it is important to consider that many patients may be entirely asymptomatic. 11 Conductive hearing loss or ear mass on examination may trigger further evaluation. Congenital cholesteatomas of the temporal bone can arise in the middle ear cleft, throughout the petrous bone, or around the cerebellopontine angle. 12 Erosion of the labyrinthine bone by CC is rare because the labyrinthine is the hardest bone in the body. Previous reports showed the rate of cholesteatoma-related semicircular canal erosion is between 6% and 8%.13,14 Erosion of the cochlea and internal auditory canal are much rarer, and our review of the literature found less than 15 cases since 1985.15-17
Previously Reported Cases of Inner Ear Erosion by Cholesteatoma.
Differential Diagnoses
The differential diagnosis for a large temporal bone lesion with extensive erosion of labyrinthine bone in a child includes rhabdomyosarcoma, cholesterol granuloma, Langerhans Histiocytosis, endolymphatic sac tumor, as well as glomus jugulare. There are key features of each pathology that allow clinicians to differentiate them from congenital cholesteatoma. Table 2 summarizes the imaging characteristics of the different erosive temporal bone lesions (Table 2). Rhabdomyosarcoma differs from cholesteatoma by the presence of contrast enhancement and lack of DWI restriction on MRI. 18 Cholesterol granuloma can be distinguished from congenital cholesteatoma by the low to intermediate level of T1-weighted signal in cholesteatoma compared to cholesterol granuloma which is hyperintense in T1 and T2. 19 Langerhans histiocytosis (LCH) and rhabdomyosarcoma demonstrate very similar MRI and CT characteristics, and differentiation often requires tissue diagnosis. LCH has a predilection for bone marrow spaces in the petrous bone. 20 Endolymphatic sac (ELS) tumors are exceedingly rare and associated with Von Hippel-Lindau disease. CT of ELS may show calcifications which may be present in cholesteatoma as well. MRI of ELS are hyperintense T1, heterogeneous enhancement, and hypergenous T2. 21 The epicenter of ELS is at the endolymphatic sac, which is unusual for CC. Glomus jugulare tumors are centered on the jugular bulb, with moth-eaten bony erosion on CT. It demonstrates on MRI low T1 and high T2 signal, marked enhancement with flow void.22-24
Imaging Characteristics of Erosive Temporal Bone Lesions MRI.
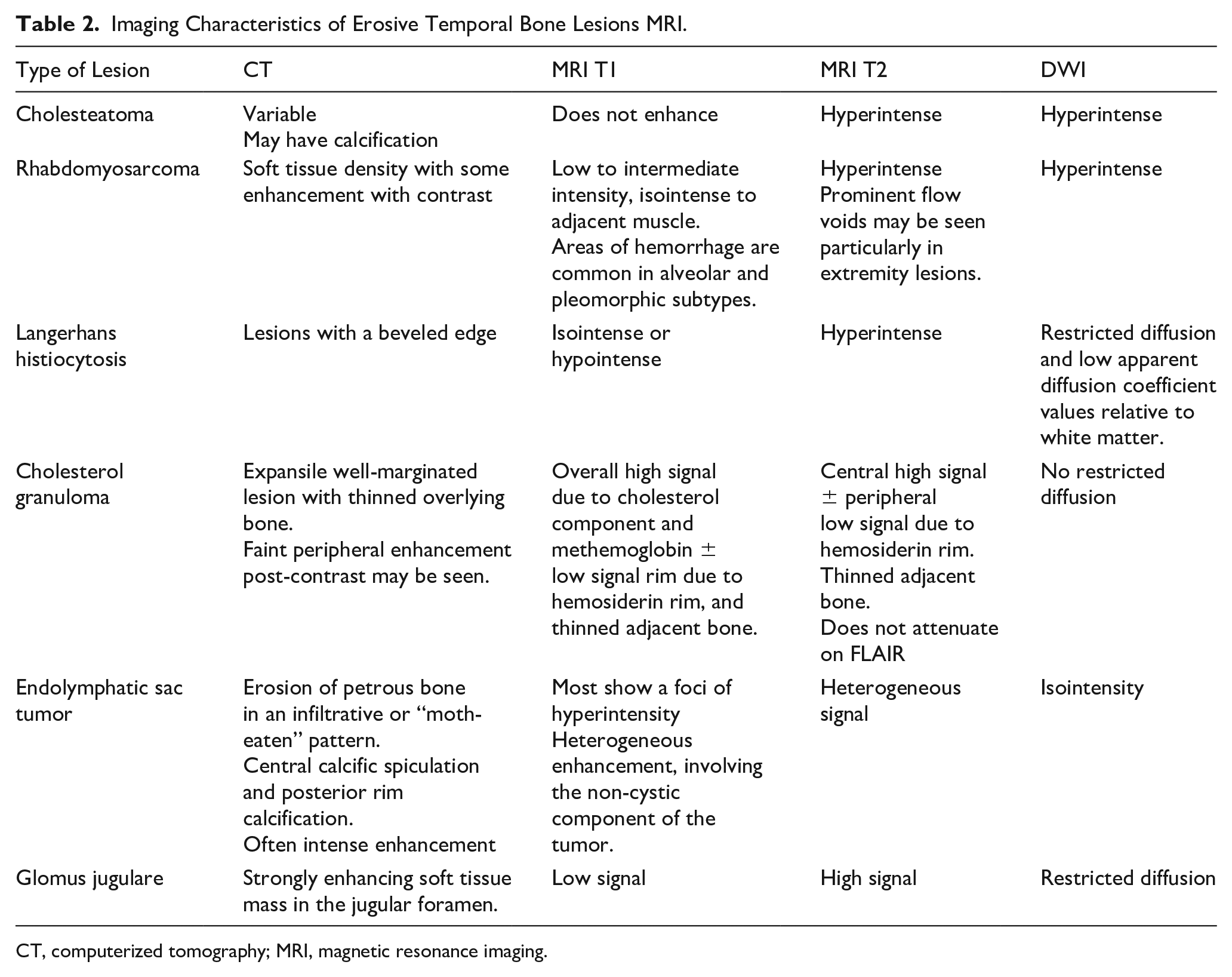
CT, computerized tomography; MRI, magnetic resonance imaging.
Management of Congenital Cholesteatoma
Surgery remains the main treatment for CC; however, there is a high recurrence rate ranging from 18% to 50%. 25 The rate of recurrence is directly related to the extent of disease at diagnosis.11,26 The incidence of congenital cholesteatomas is on the rise as a result of increasing awareness and reporting. 7 Rarely is a canal wall-down procedure needed in CC. 27 In this case, despite detection at a very young age, the disease was already advanced at presentation. Extensive disease requires careful evaluation of risks for meningitis, CSF leak, and intracranial extension. In very young patients such as this, surgical management also weighed the limited surveillance options because MRI and/or mastoid cavity debridement require sedation at this age.
Superimposed bacterial and/or fungal infection was treated aggressively to prevent intracranial extension of infection. The report also suggests that bacterial and fungal infections may promote bone erosion by cholesteatoma.28,29
Conclusion
This case is unique in the complete lateral displacement of the cochlea and aggressive erosion of the internal auditory canal and lateral semicircular canals in a very young child. It serves to remind otolaryngologists of the silent yet infiltrative nature of congenital cholesteatoma. The very young age of the patient and the presence of otorrhea at 1 month of age raise the question of whether this lesion could have been developing in utero. It lends support to the theory of developmental epithelial rests in congenital cholesteatoma. As surgeons, we need to consider whether to treat these very early congenital cholesteatomas more like tumors for their very aggressive behavior.