
Abstract
Malignant rhabdoid tumor (MRT), first described by Beckwith and Palmer in 1978 as a variant of Wilms’ tumor, is an aggressive neoplasm with early onset and poor prognosis. Malignant extra-renal rhabdoid tumors (MERTs) can arise in various locations and often present at birth as a disseminated disease without an identifiable primary site, posing significant diagnostic and therapeutic challenges. This case highlights the complexity of diagnosing an 11-day-old preterm infant with multiple enlarging masses, including a hypervascular forehead lesion, parotid mass, and soft tissue masses in the leg and hip. Initial imaging, pathology, and immunohistochemistry findings were inconclusive, necessitating advanced molecular studies, which ultimately confirmed MRTs through the loss of SMARCB1 expression. However, the diagnosis was delayed due to overlapping differential considerations, including fibrosarcoma and other sarcomas. Despite extensive multidisciplinary efforts and tailored treatments, the tumors progressed aggressively, emphasizing the challenges in diagnosing and managing such rare conditions. Given their complexity, MERTs require a comprehensive diagnostic approach combining clinical evaluation, histopathological analysis, and molecular studies, with a high level of clinical suspicion essential for timely treatment.
Keywords
Introduction
Malignant rhabdoid tumor (MRT) was first described by Beckwith and Palmer in 1978 as a variant of Wilms’ tumor, initially identified in the kidney. It is distinguished by its unique histological morphology, early onset within the first year of life, and highly aggressive clinical course, with long-term survival being exceptionally rare. 1 Malignant extra-renal rhabdoid tumors (MERTs) are known to originate in various anatomical sites, including deep axial regions, extremities, cutaneous tissues, and visceral organs. 2 When diagnosed prenatally, MERTs often emerge at birth or shortly thereafter as a widely disseminated disease, without any identifiable primary tumor, adding to its enigmatic and aggressive nature. 3 The diagnosis of MERT is complex, relying on histopathology, immunohistochemistry, and molecular studies for accurate confirmation. 4 The SWItch/Sucrose Non-Fermentable (SWI/SNF) complex is a chromatin-remodeling system crucial for gene regulation, composed of the core subunit SMARCB1 (INI1) and catalytic subunits SMARCA2 (BRM) and SMARCA4 (BRG1). 5 Loss of SMARCB1 protein expression on chromosome 22q11.2 is observed in over 80% of MERT, with these alterations occurring either as familial mutations or more commonly as de novo events. 1 Treatment of MERT is challenging due to the absence of national guidelines, with the approach depending on the initial diagnosis and physician preference. 2
Case Presentation
This is the case of an 11-day-old preterm female infant, delivered at 33 weeks of gestation through Cesarean section. The parents are non-consanguineous and have 3 other healthy children. The infant was transferred from an external facility for further evaluation and management of enlarging masses on the forehead, left cheek, and right lower leg. A prenatal ultrasound conducted at 32 weeks of gestation had detected a frontal mass, but no other abnormalities were identified. At birth, the infant had an Apgar score of 8 at 1 minute and 9 at 5 minutes.
Upon admission to our facility, the patient was stable, maintaining adequate oxygen saturation on room air and feeding well. Physical examination revealed a large, pedunculated, well-vascularized mass on the forehead. The mass was predominantly soft with some firmer regions, exerting a mass effect that partially covered the left eye and compressed the left nostril. A similar mass was seen in the left parotid region, and on the right leg Figure 1a. Further evaluation revealed the presence of a small ventricular septal defect, while the remainder of the examination was unremarkable.
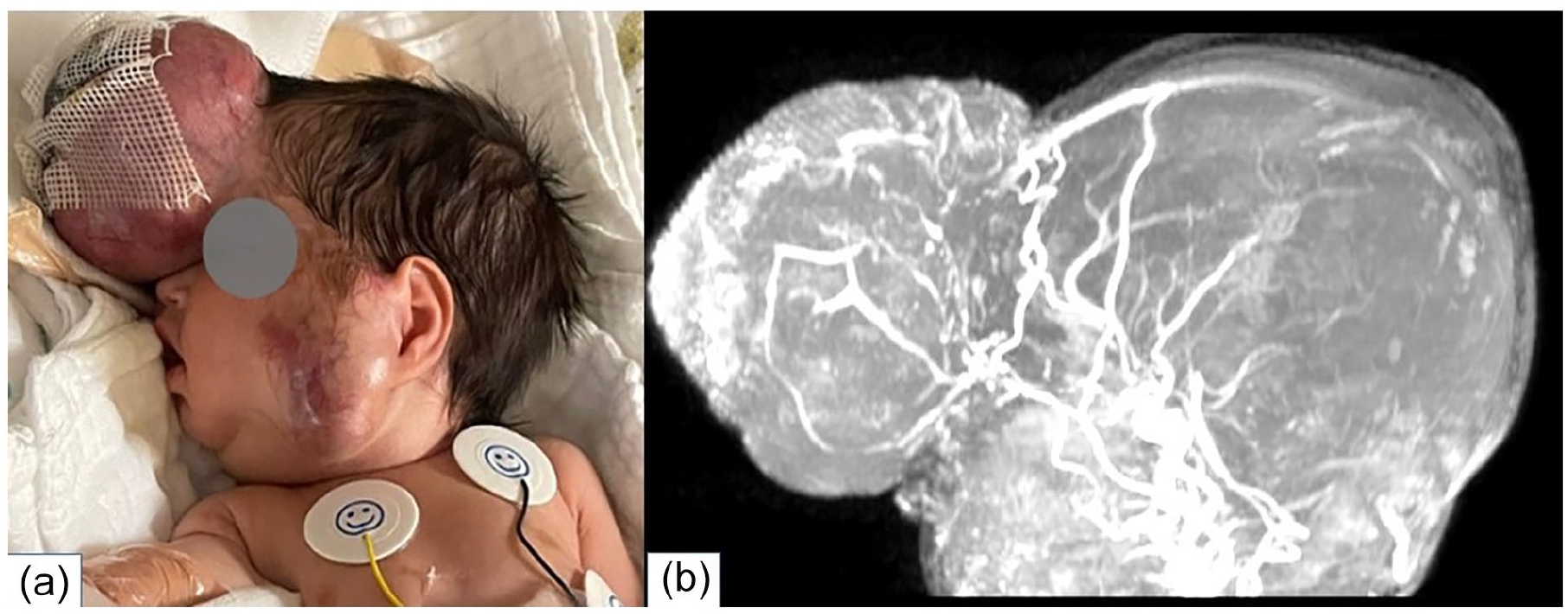
(a) An image of the patient shows a large forehead lesion covered with Bactigras due to oozing, alongside a left parotid mass with prominent subcutaneous vascularity. (b) High-resolution MRI with MIP and sagittal MRV reveals a large right-sided draining vein emptying into the right superior ophthalmic vein. MRI, magnetic resonance imaging; MIP, Maximum Intensity Projection; MRV, Magnetic resonance venography.
Initial laboratory tests revealed an elevated white blood cell count of 15,900 and a C-reactive protein level of 1.8. Hemoglobin was low at 9.7 g/dL. The coagulation profile showed prolonged prothrombin and partial thromboplastin times, elevated D-dimer levels, and a significantly reduced fibrinogen level. Platelet levels were normal, carbon dioxide was markedly low, while bilirubin and lactate dehydrogenase levels were mildly elevated.
A computed tomography (CT) scan of the facial bones, along with whole-body magnetic resonance imaging (MRI), Magnetic resonance angiography (MRA), and Magnetic resonance venography (MRV), revealed a large, hyper-vascular subcutaneous forehead mass measuring 7.4 × 4.9 × 5.9 cm. It exhibited prominent peripheral vessels, internal bleeding, and a solid peripheral component surrounding a central area of necrosis, hemorrhage, or calcifications. The mass did not extend into the brain parenchyma or orbit. Branches of the superficial temporal, supra-trochlear, and angular arteries encircled the mass superficially, with multiple vessels traversing within the tumor. A large draining vein on the right side empties into the right superior ophthalmic vein and another on the left drains into the left external jugular vein Figure 1b. No arteriovenous malformation was identified. The left parotid space lesion, measured 4.6 × 2.5 × 2.8 cm, had similar characteristics. There are bilateral level II masses, suggestive of metastatic lymph nodes. Additional findings included an oval-shaped soft tissue mass in the anterior aspect of the distal right leg (1.6 × 1.2 cm) and a 1.5 cm enhancing intramuscular mass in the lateral aspect of the left hip/proximal left thigh. The differential diagnosis includes fibrosarcoma, atypical teratoid rhabdoid tumors, or other types of sarcoma.
Due to the progressive growth of all the masses, a multidisciplinary team comprising specialists from pediatric intensive care, pediatric hematology and oncology, otorhinolaryngology, and plastic surgery decided to proceed with an excisional biopsy of the forehead lesion to establish a definitive diagnosis and address the disfiguring effects.
During surgery, dissection was carried down to the bone, and notably, the tumor easily separated from the underlying forehead bone. Bleeding from the supratrochlear vessels was controlled with compression, bone wax, and Floseal and cautery. The wound was closed with a silver dressing, and the patient was transported intubated to the neonatal intensive care unit.
Initial pathology revealed a subepidermal vascular tumor characterized by dilated vascular channels and a predominantly round-to-oval cell infiltrate with abundant cytoplasm, surrounded by prominent necrosis. The tumor cells displayed large nuclei with clearing, prominent nucleoli, and brisk mitotic activity, indicative of an undifferentiated malignant round-cell tumor. Immunohistochemistry was performed for further characterization, showing weakly positive staining for CD99, CK AE1/3, SATB2, and SALL4 (Figure 2a). The tumor was negative for synaptophysin, desmin, CD34, CD45, FLi-1, SOX10, CD1a, SMA, WT1, inhibin, and HMB45.
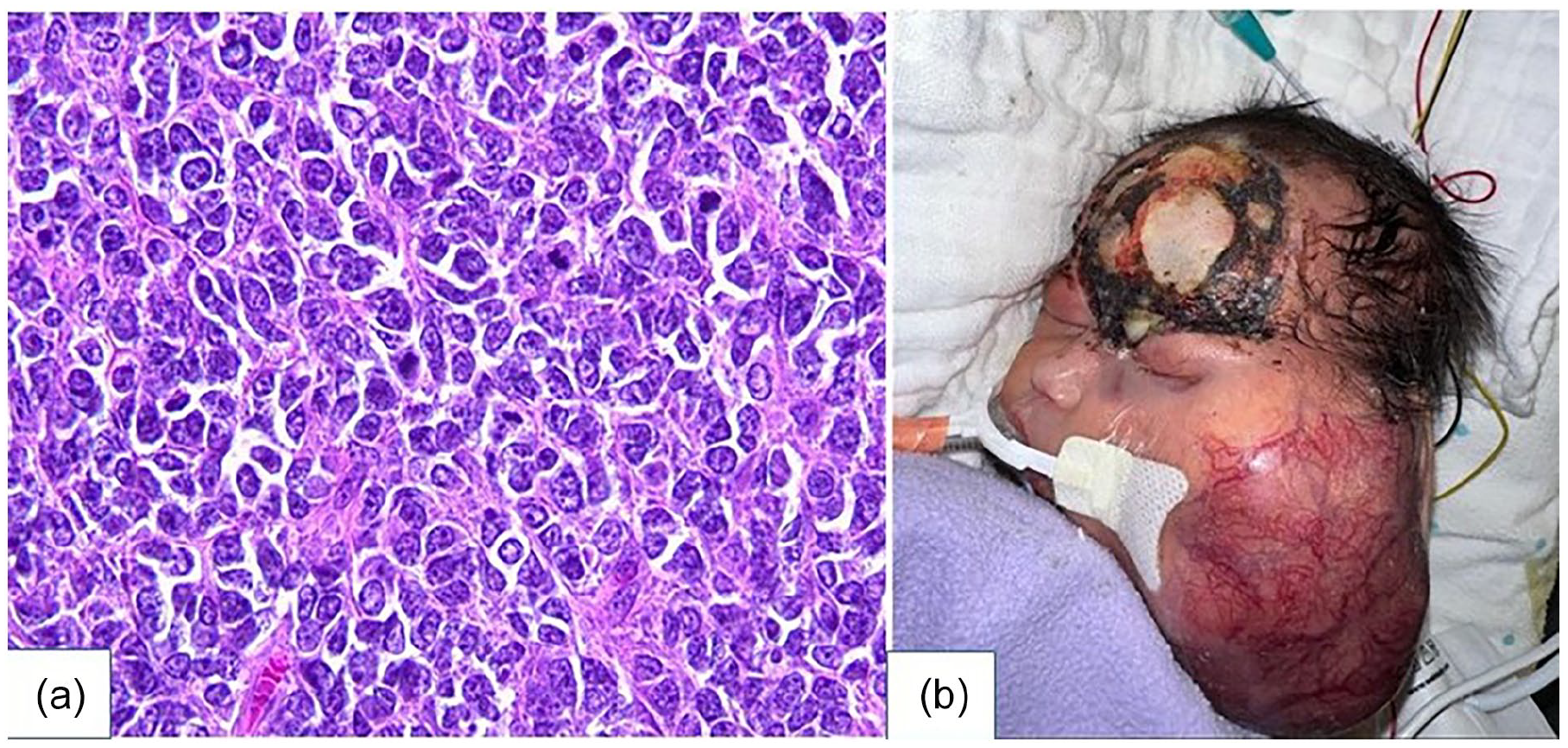
(a) Histologic examination reveals a high-grade malignant neoplasm composed of oval to round tumor cells with moderate to abundant cytoplasm, large nuclei with prominent clearing, conspicuous nucleoli, and brisk mitotic activity. (b) Postoperative image, taken 1 week before passing, shows a rapid enlargement of the left parotid mass without forehead mass regrowth.
For the first few days after surgery, the patient remained clinically unstable, leading to a delay in initiating chemotherapy for nearly 7 days. Once stable, she was initially managed as a case of infantile fibrosarcoma or infantile myofibromatosis while awaiting results from advanced molecular studies conducted abroad. The standard treatment typically includes chemotherapy with vincristine, dactinomycin, and cyclophosphamide along with complete surgical resection. However, due to the risk of hepatopathy, the decision was made to initiate cyclophosphamide alone at 40 mg/kg based on the rhabdomyosarcoma protocol, but no improvement was observed. Targeted therapy with larotrectinib was subsequently added, followed by vinblastine and a reduced dose of methotrexate. Despite these efforts, the tumors exhibited rapid progression (Figure 2b), leading to the patient’s clinical deterioration, marked by escalating respiratory distress, generalized edema, and renal failure.
Given the poor prognosis, the clinical situation was discussed with the parents, who consented to a do-not-resuscitate status. The patient experienced progressive bradycardia and desaturation, ultimately resulting in cardiac arrest, 1 month after admission.
Molecular testing revealed a loss of SMARCB1 (INI-1) expression with retained SMARCA4 (BRG-1) expression, and negative FISH staining for ETV6 gene rearrangement, findings consistent with atypical teratoid/rhabdoid tumors or MRTs.
Discussion
Rhabdoid tumors are rare and aggressive malignant tumors of the pediatric population. The exact incidence of MRTs is uncertain; however, a European study by Reinhard et al estimates it to be between 0.1 and 0.5 cases per million children per year. 6
Rhabdoid tumors exhibit a bimodal distribution, primarily affecting neonates and infants during the first year of life, but occasionally occurring in older individuals. In older infants and children, these tumors typically arise from the kidneys or central nervous system (CNS) and are generally localized. By contrast, fetuses and neonates more commonly present with tumors originating from extra-renal and extra-CNS sites.3,6 Metastases commonly involve the skin, placenta, bones, lungs, lymph nodes, skeletal muscles, and liver, resulting in rapid progression and early mortality.2,7 Survival rates vary in the literature, Isaacs reported a 2.3% in patients with metastatic MERT. 3 Sultan et al reported 229 cases, including 40 pediatric nonrenal and noncranial rhabdoid tumors, with a survival rate of 20% in children under 2 years, 8 whereas Reinhard et al observed a 30% survival rate in 25 children with similar pathology. 6
Physicians managing MRTs face dual challenges: ensuring diagnostic accuracy and developing effective treatment strategies. Diagnosis of extrarenal rhabdoid tumors is a multifaceted process requiring a comprehensive approach that integrates clinical, histopathological, immunohistochemical, and molecular findings. Grossly the tumors are typically bulky and lobulated, appearing hypo-attenuating on CT, hypointense on T1-weighted, and heterogeneously hyperintense on T2-weighted MRI often with necrotic areas. However, these imaging characteristics are nonspecific, necessitating confirmation through comprehensive pathology studies. 4 Histologically, rhabdoid tumor cells are characterized as large, round, blue cells with abundant eosinophilic cytoplasm, eccentric vesicular nuclei, and prominent nucleoli—hallmark features of rhabdoid cells. These cells are typically arranged in sheets, clusters, or single-file patterns. In addition, some cells may display spindled or primitive small-cell morphology, with most tumors exhibiting a high mitotic rate. However, the morphological features of rhabdoid tumors are not exclusive and can overlap with other malignancies, potentially leading to diagnostic errors, as demonstrated in our case and similarly in a reported case by Ramieri et al, of a frontonasal congenital rhabdoid tumor initially misdiagnosed as esthesioneuroblastoma based on pathology.1,4,9,10
The immune profile of MRTs is characterized by keratin positivity and loss of INI1 expression due to biallelic loss of the SMARCB1 gene on chromosome 22q11.2.4,8 In tumors with MRT-like features but retained SMARCB1 expression, testing for SMARCA4 protein loss is necessary, as these tumors belong to the distinct subgroup of SMARCA4-mutated MRTs.1,5 Most rhabdoid tumors arise from somatic loss of both SMARCB1 alleles, while 15%-30% are associated with germline mutations or deletions which typically acquired de novo which could be the case of our patient who had both healthy parents and siblings. Strikingly, SMARCB1 loss is not exclusive to rhabdoid tumors and has been identified in other tumors, particularly proximal epithelioid sarcomas. 9
There are no standardized treatment guidelines for MERTs, and the literature on this is highly controversial. However, Bourdeaut et al found that MERT shares similar chemosensitivity and recurrence patterns with renal and cerebral rhabdoid tumors. 2 Multimodal therapy, with surgery as the cornerstone and adjuvant chemotherapy and age-based radiotherapy, has shown promise in improving survival. A systematic review and meta-analysis by Andres et al demonstrated the efficacy of sarcoma-targeting protocols, typically alkylator-based regimens such as cyclophosphamide, carboplatin, or cisplatin, combined with methotrexate, etoposide, or vincristine, which aligns with the treatment approach used for our patient. 11
Hence, the integration of clinical presentation with multiple diagnostic modalities highlights the critical need for a high index of suspicion and meticulous clinical judgment in evaluating rapidly growing tumors, as every detail can be pivotal in achieving an accurate diagnosis. This approach can help mitigate delays in initiating appropriate treatment, whether as salvage therapy or adjuvant treatment following surgery.
Conclusion
MERTs present significant diagnostic challenges due to their nonspecific imaging characteristics, overlapping histopathological features with other malignancies, and complex molecular profiles. Accurate diagnosis requires a systematic approach integrating clinical evaluation, histopathological analysis, and molecular studies, with a high level of clinical suspicion essential to ensure timely and appropriate treatment initiation.