
Abstract
To determine the genetic causes of sensorineural hearing loss (SNHL) associated with inner ear anomalies, 11 unrelated Turkish individuals diagnosed with SNHL and an inner ear anomaly using temporal bone computed tomography and inner ear magnetic resonance imaging underwent exome or whole genome sequencing to identify underlying genetic defects. None of the individuals was diagnosed with a recognized syndrome. Four of the 11 probands were homozygous for SLC26A4 variants, c.283G>A, c.845G>A, c.1061T>C, and c.1198delT. Another proband was homozygous for a TECTA variant, c.4163G>A. Patients with variants of the SLC26A4 gene had bilateral enlarged vestibular aqueduct, bilateral incomplete partition type 2 anomaly, bilateral hypoplastic cochlea and bilateral enlarged vestibular aqueduct plus hypoplastic cochlea anomaly. Patients with the variant TECTA gene had bilateral hypoplastic cochlea. This study identified variants of SLC26A4 in 36% of probands with inner ear anomalies. While we identified a variant of the TECTA gene in a proband with cochlear hypoplasia, further studies are needed to see if TECTA variants can cause cochlear malformations.
Introduction
Hearing loss is a common sensory disorder in humans. The prevalence of permanent, moderate, and severe sensorineural hearing loss (SNHL) is estimated to be between 1 and 3 for every 1000 live births. 1
Studies show that more than 50% of congenital or prelingual SNHL is caused by genetic factors. 1 Although there are no additional findings in most cases of SNHL, it has been identified as another finding in more than 400 genetic syndromes. Genetic or environmental factors can alter the development of the inner ear, leading to abnormalities. In previous studies, abnormalities in the inner ear were detected in 7.5% to 21% of patients with SNHL.2-5
Twenty percent of those with SNHL also have an inner ear defect. Even though the first accounts are from the 18th century, our understanding of these situations has come a long way in the last few decades. A rehabilitation alternative is available for the majority of these conditions at the moment. Surprisingly, we know little about what causes these abnormalities. Specifically, new information regarding the probable association between hereditary abnormalities and inner ear deformities has been made available by the development of genetics. More recently, an inner ear abnormality has been linked to the well-known occurrence of SNHL in syndromic conditions. These abnormalities may be regarded as hallmarks of the condition in certain instances.2-5
Determining the genetic basis for inner ear anomalies can facilitate the development of future treatments. Proper genetic counseling is also necessary for affected families. However, only a few studies were conducted on this issue.6,7 This study aims to provide data on our cohort’s genetic causes of inner ear anomalies.
Materials and Methods
This study was approved by the Dicle University Medical School Ethics Committee (Turkey), the Ankara University Medical School Ethics Committee (Turkey), and the University of Miami Institutional Review Board (USA). A signed informed consent form was obtained from each participant or, in the case of a minor, from the parents.
In this study, we included 11 probands with inner ear anomalies. In adult participants, pure tone audio audiometry and, when necessary, auditory brainstem response (ABR) were applied; in pediatric participants, pure audio audiometry, autoacoustic emission, ABR, free audiometry, tympanometry, and stapes reflex were used, and diagnosis of SNHL was established. Temporal bone computed tomography and inner ear magnetic resonance images were obtained in all affected participants.
Blood samples were taken from individuals with inner ear anomalies and, when available, from their parents. Exome or genome sequencing was performed as reported previously.8,9 DNA variants (single nucleotide, indel, and copy number variants) mapping to the coding regions of genes and 20 bp within introns were evaluated. All previously recognized deafness genes were retrieved from the Hereditary Hearing Loss Homepage (http://hereditaryhearingloss.org/), and Online Mendelian Inheritance in Man (http://omim.org/) were filtered and interpreted under autosomal dominant, recessive, X-linked inheritance models. The guidelines of the American College of Medical Genetics and suggestions from the ClinGen Hearing Loss Gene Curation Expert Panel were considered for variant interpretation (PMID: 25741868, PMID: 30245029, PMID:26226137). Candidate variants were confirmed by Sanger sequencing.
Results
We evaluated 11 probands with inner ear anomalies. Seven (63%) of these patients were male and 4 (37%) were female. The mean age of these patients was 1 and 20 years. Deep bilateral SNHL was detected in all probands. The mean hearing thresholds were 85.71 ± 9783 dB in the right ear and 88.18 ± 6645 dB in the left ear. All patients had inner ear malformation in both ears (Table 1). Inner ear anomalies detected by imaging techniques were classified according to the literature (Table 2).10,11
Distribution of Probands.
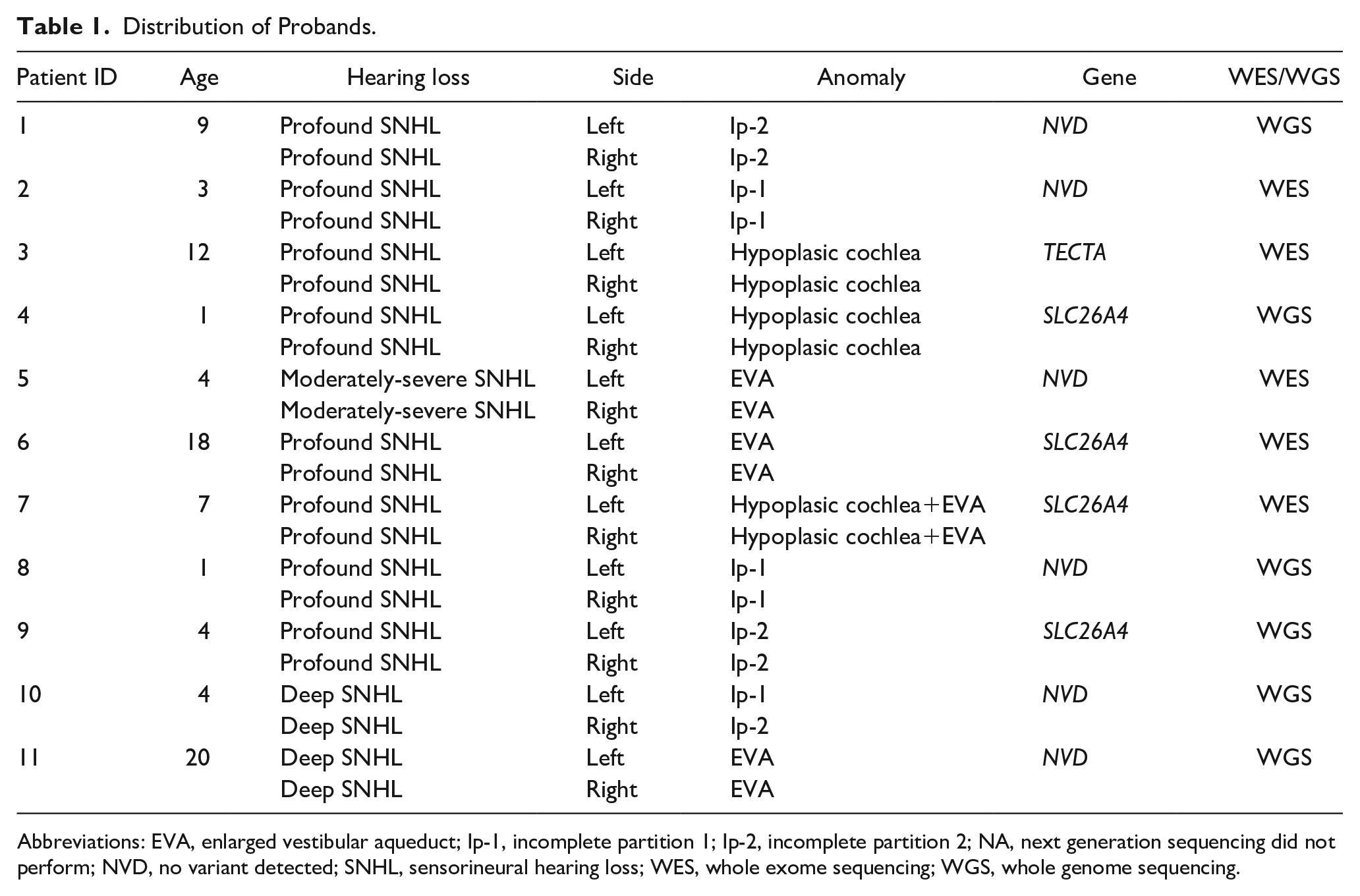
Abbreviations: EVA, enlarged vestibular aqueduct; Ip-1, incomplete partition 1; Ip-2, incomplete partition 2; NA, next generation sequencing did not perform; NVD, no variant detected; SNHL, sensorineural hearing loss; WES, whole exome sequencing; WGS, whole genome sequencing.
Radiological Results on Detected Inner Ear Malformations.
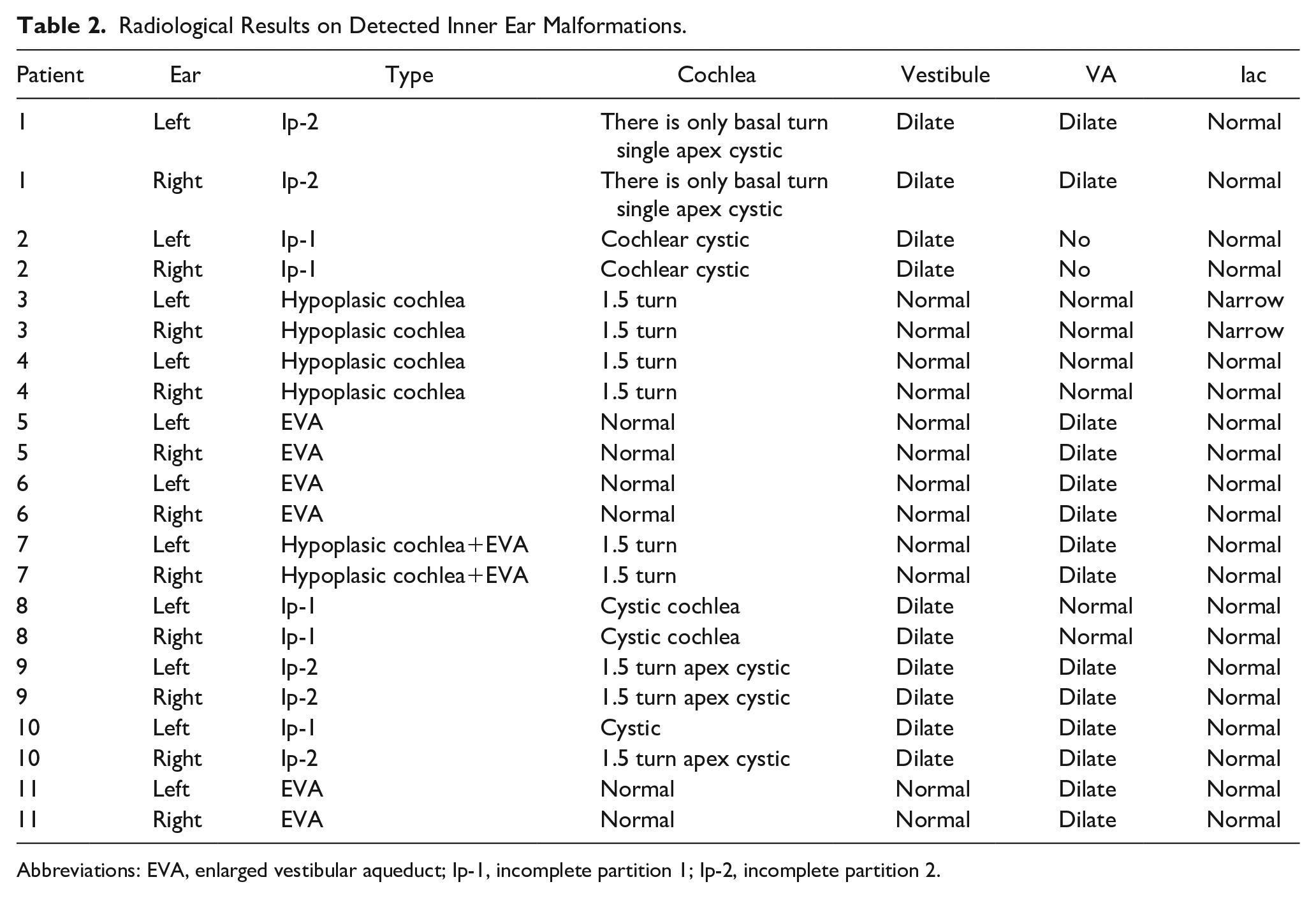
Abbreviations: EVA, enlarged vestibular aqueduct; Ip-1, incomplete partition 1; Ip-2, incomplete partition 2.
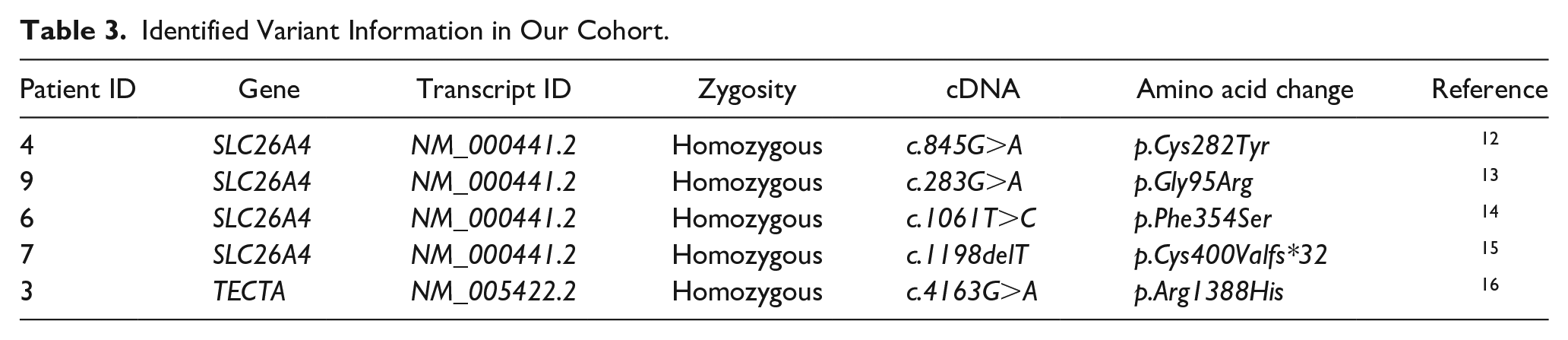
SLC26A4 gene variants were detected in 4 cases, and a variant of the TECTA gene associated with non-syndromic hearing loss was detected in a patient with hypoplastic cochlea. In this study, 4 variants (c.283G>A, c.845G>A, c.1061T>C, and c.1198delT) identified in SLC26A4, which were believed to be associated with inner ear abnormalities, were detected in 4 patients. One of these 4 patients with the SLC26A4 gene mutation had a bilateral hypoplasic cochlea, a bilateral enlarged vestibular aqueduct, and a Mondini type 2 bilateral incomplete partition anomaly. A hypoplasic cochlea anomaly and a bilateral enlarged vestibular aqueduct were observed in a patient. The bilateral hypoplasic cochlea was detected in our patient with the TECTA gene mutation (Table 3).
Identified Variant Information in Our Cohort.
Discussion
Inner ear anomalies can be isolated or can occur as part of other syndromes involving other systems. While many syndromes have been associated with inner ear anomalies in humans, 6 no additional findings were found in our patients, making their diagnosis non-syndromic hearing loss. Mutations in more than 110 genes today have been associated with non-syndromic hearing loss.12,13
Among all patients with non-syndromic hearing loss, several genes have been discovered that cause inner ear anomalies, including SLC26A4, POU3F4, COCH, ROR1, and FOXF2.6,14 SLC26A4 gene mutations have been found in people with non-syndromic autosomal recessive deafness associated with vestibular aqueduct enlargement and Ip-2 anomaly. 17 This gene encodes an anion transporter protein called pendrin. It has been determined that this gene is active in the endolymph reabsorption region of the cochlea in the inner ear, and the endolymph homeostasis is disrupted due to its mutations, leading to hearing loss. The gene is located in the 7q31 region and consists of 21 exons. More than 200 mutations have been reported so far. These gene mutations may be associated with sporadic and familial forms of Pendred syndrome or vestibular aqueduct enlargement associated with non-syndromic SNHL.14,15 No additional pathologies of Pendred syndrome were observed in any of our patients. Symptoms and findings of this syndrome may appear later in life.14,15 Therefore, these patients should be followed up concerning the thyroid gland that may be seen in this syndrome.
Temporal bone anomalies such as Mondini dysplasia, enlarged vestibular aqueduct, and cochlear spiral duct hypoplasia are seen in mutations in the SLC26A4 gene. Campell et al 7 detected SLC26A4 gene mutations in 5 of 6 families with enlarged vestibular aqueduct and 4 of the families with Mondini anomaly. 7 Of our patients, we detected the SLC26A4 gene. A bilateral enlarged vestibular aqueduct was observed in 1 bilateral hypoplasic cochlea bilateral enlarged vestibular aqueduct in 1, a bilateral incomplete partition type 2 Mondini was observed in 1, and a bilateral enlarged vestibular aqueduct coexisting with cochlear hypoplasia was observed in 2.
Regarding mutations in the SLC26A4 gene, the most frequent mutation was c.716T>A transformation in a study conducted in Pakistan. The most frequent mutation was c.1001+1G>A mutation in a survey conducted with the Caucasian population. The most frequent mutations were c.919-2A>G, and c.2168A>G in individuals of East Asian origin.16,17 Mutations that were different from these have been detected in our patients. This shows that there are many different mutations in different ethnic populations.16,17
In this study, 4 variants (c.283G>A, c.845G>A, c.1061T>C, and c.1198delT) identified in SLC26A4 were detected in 4 patients. Of our patients, homozygous c.283G>A mutation in the third exon of the SLC26A4 gene SLC26A4 was detected in our patient found to have a bilateral Mondini anomaly, homozygous c.845G>A mutation in the seventh exon of the SLC26A4 gene was detected in our patient found to have bilateral hypoplasic cochlea, homozygous c.1061T>C mutation in a ninth exon of gene SLC26A4 was detected in our patient found to have bilateral enlarged vestibular aqueduct, homozygous c.1198delT mutation in a seventh exon of the gene SLC26A4 was detected in our patients who are siblings and found to have bilateral hypoplasic cochlea and enlarged vestibular aqueduct.18,19
Mutations in the POU3F4 gene may be associated with IP-III (missing partition type 3), cochlear hypoplasia, and stapes fixation (DFN3). 20 Considering the literature, mutations in the COCH gene have been found in patients with semicircular canal dehiscence, stenosis, and enlarged vestibular aqueduct.20-22 Based on these findings, COCH gene mutations are one of the possible inherited autosomal dominant causes of inner ear anomalies. A mutation in the ROR1 gene was detected in 2 children of a family with non-syndromic congenital autosomal recessive SNHL with a joint cavity anomaly. 23 In our study, no mutations in the POU3F4, COCH, ROR1, or FOXF2 gene were detected that were thought to be associated with inner ear abnormalities.
A missense mutation in the TECTA gene was detected using the whole genome sequencing method in a patient with bilateral hypoplastic cochlea. In our patient, homozygous c.4163G>A transition in the TECTA gene was identified. This gene encodes a protein called the a-sector, the fundamental non-collagen component of the tectorial membrane. 24 The tectorial membrane is connected to specialized hair cells in the inner ear. This gene is located in the 11q23-25 chromosome region and has 23 exons. This gene mutation can cause both autosomal recessive and autosomal dominant hearing loss associated with non-syndromic hearing loss. So far, 13 different mutations have been determined in this gene.24-27 The TECTA gene mutation has not previously been found to be associated with inner ear abnormalities. There may be another gene that explains the inner ear anomaly phenotype.
As a result, no gene mutation was detected in 7 patients in this study. In our study, mutations were detected in less than half of the cases in which we found abnormalities in the inner ear. Among these mutations, we detected that the SLC26A4 gene-related mutations c.283G>A, c.845G>A, c.1061T>C, and c.1198delT are associated with inner ear anomalies.
This study is essential in determining the genetic structure of inner ear anomalies and is a step toward possible gene therapies. Strengthening this step requires increasing the number of similar studies.
Footnotes
Acknowledgements
NIH R01DC009645 and R01DC012836 supported this work in MT.
Author Contributions
Umit Yilmaz: Planning, designing, conducting literature surveys, and providing active intellectual support. Muzeyyen Yildirim Baylan: Planning, designing, literature survey, statistical analysis, writing, active intellectual support, submission. Duygu Duman: Planning, designing, literature survey, active intellectual support. Claire Sineni: Planning, designing, literature survey, active intellectual support. Güney Bademci: Planning, designing, literature survey, active intellectual support. Bilal Sizer: Planning, designing, literature survey, active intellectual support. Mustafa Tekin: Planning, designing, literature survey, data collection, active intellectual support, English editing.
Data Availability Statement
The available data of the study may be sent upon request for scientific purposes.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Statement
This study was approved by the Dicle University Medical School Ethics Committee (Turkey), the Ankara University Medical School Ethics Committee (Turkey), and the University of Miami Institutional Review Board (USA).
Informed Consent
Informed consent was obtained from the patients and/or their parents before they were included in the study.
Trial Registration Number/Date
Not applicable.
Grant Number
Not applicable.