
Abstract
Low-grade fibromyxoid sarcoma (LGFMS) represents an exceptionally rare soft-tissue tumor, challenging to diagnose, and notorious for relentless recurrence and proliferation postsurgical resection. Primary symptoms of LGFMS include nasal congestion and rhinorrhea, accompanied by cheek numbness and distension. In this article, we report the diagnosis and treatment of a case of low-grade LGFMS originating in the maxillary sinus (MS). A 64-year-old male diagnosed with LGFMS of the left MS, undergoing 3 surgeries over a 1-year period with subsequent local recurrence. Following inconclusive postoperative pathology after the initial surgery, the patient experienced recurrence 2 months postsurgery, necessitating a second operation, which confirmed the LGFMS diagnosis pathologically. Radiation therapy commenced 1 month after the second surgery; however, recurrence transpired 6 months later, leading to a third operation. Subsequently, recurrence occurred again 8 months post third surgery, with the patient currently undergoing targeted therapy. This case underscores the distinct characteristics and therapeutic challenges inherent in LGFMS through the narrative of diagnosis and progression of LGFMS originating in the MS.
Introduction
Low-grade fibromyxoid sarcoma (LGFMS), an uncommon form of soft-tissue sarcoma, typically manifests in the deeper layers of the soft tissues in the distant limbs. 1 Despite its benign histological appearance, LGFMS exhibits aggressive behavior. 2 While it primarily afflicts young adults, occurrences in children and older adults are not uncommon. 3,4 Accurate diagnosis hinges on postoperative pathological analysis. Wide local excision stands as the primary treatment option due to LGFMS’s minimal mitotic activity, rendering chemotherapy and radiotherapy largely ineffective. 5 Despite its slow growth and protracted clinical course, LGFMS poses a substantial risk of local recurrence and distant metastasis. 2 Our report highlights a case of recurrent LGFMS identified in the maxillary sinus (MS) following 3 extensive surgical excisions and 1 course of radiotherapy over a 1-year period.
Case Report
A 64-year-old male patient was admitted to the hospital reporting left-sided nasal congestion, rhinorrhea, and intermittent epistaxis persisting for over 3 months. During physical examination, a soft-tissue mass obstructing the middle meatus of the left nasal passage was observed. Endoscopic examination revealed a natural nasopharynx, and routine blood tests demonstrated normal results. Enhanced computed tomography (CT) and magnetic resonance (MR) scans of the paranasal sinus revealed contrast-enhancing soft-tissue masses in the left MS, ethmoid sinus, and middle nasal canal, extending into the left nasal passage and filling the middle meatus (Figures 1 and 2).
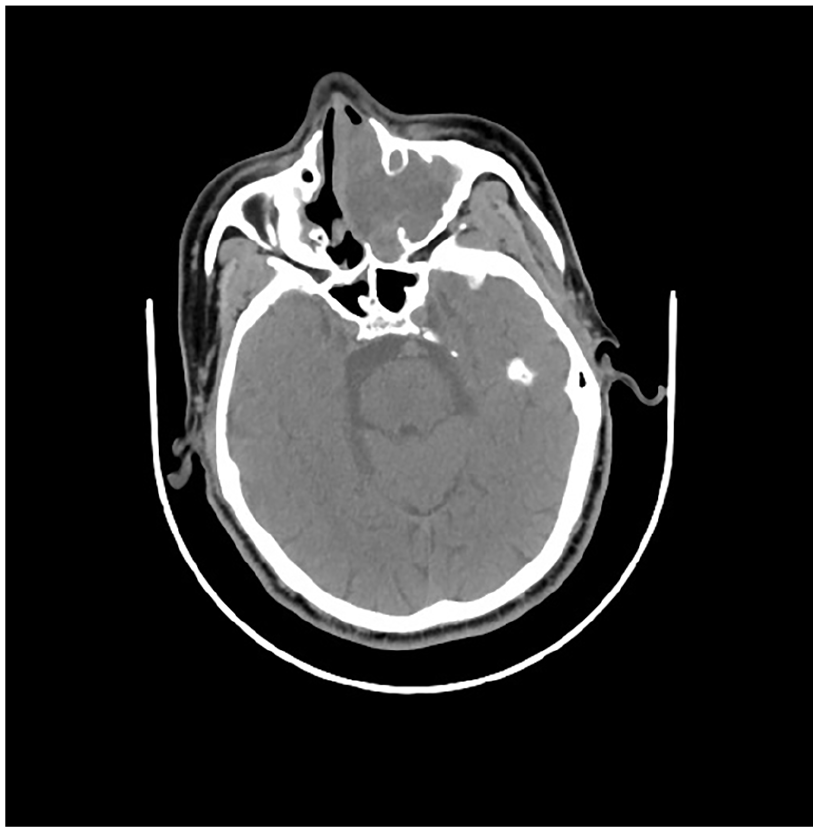
Axial view of preoperative CT imaging.
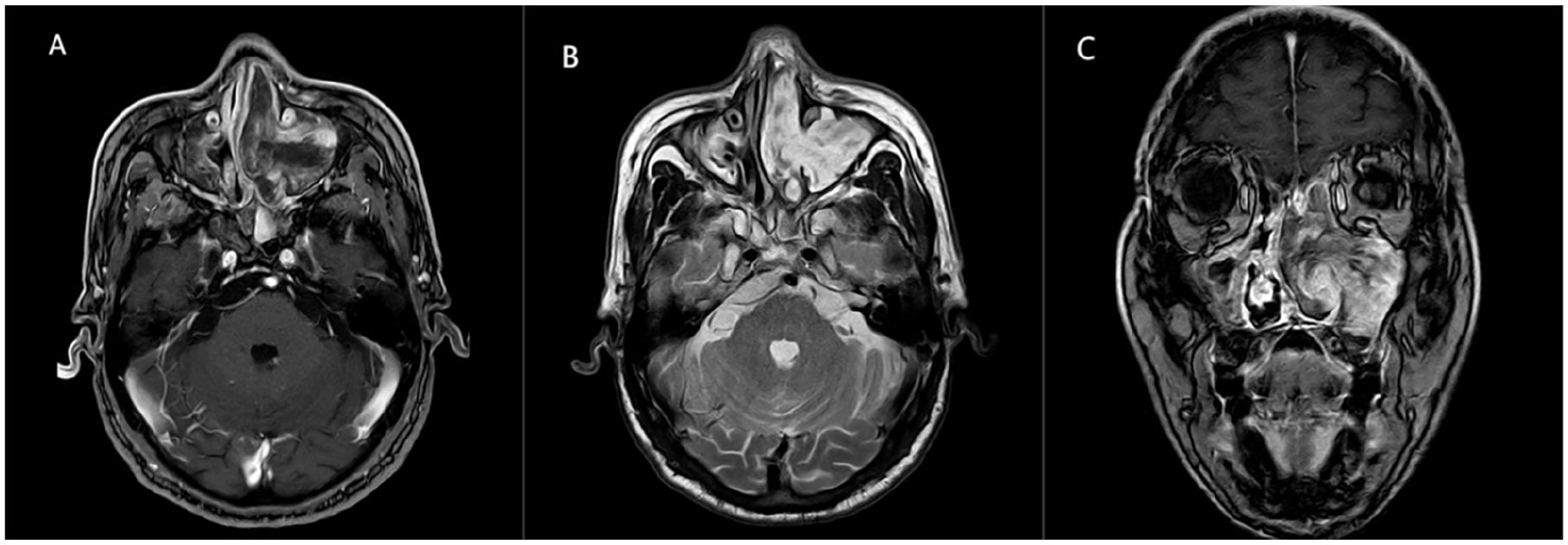
Preoperative MRI scans depicting the tumor mass within the left MS. (A) TI-weighted images. (B, C) Both T2-weighted images. MS, maxillary sinus.
No signs of metastasis or invasion were noted. The patient underwent a left anterior lacrimal recess approach, along with resection of the left MS tumor and partial resection of the left maxilla, all performed under general anesthesia. Histopathological analysis indicated a tumor of mesenchymal origin. Immunohistochemical examination revealed positive outcomes for vimentin but negative outcomes for desmin, CD34, S100, Mucin-4, AE1/AE3, and SMA, with a Ki-67 proliferation index of 25%. Despite efforts, the tumor remained undiagnosed initially.
Two months later, the patient returned to our department experiencing left nasal congestion accompanied by cheek pain and numbness. Physical examinations demonstrated a recurrent soft-tissue mass obstructing the middle meatus in the left nasal passage, along with pain and swelling on the left side of the face (Figure 3). CT and MR scans displayed a contrast-enhancing soft-tissue mass in the left MS, ethmoid sinus, and middle nasal canal, eroding bone structures and causing destruction of the left nasal passage and MS anterior walls (Figures 4 and 5). No obvious contraindications were found, and a second operation was performed under general anesthesia. The surgical methods involved endoscopic resection of the left nasal cavity and sinus tumor and partial resection of the maxilla. During the operation, complete loss of the maxillary anterior wall bone structure was observed due to the soft-tissue mass filling the MS, with tumor invasion into the orbital cavity and facial adipose tissue. Macroscopic tumor removal was achieved. Pathological examination revealed tumor cells with atypia, enlarged nuclei, angular irregular morphology, mitotic figures, and extensive mucous degeneration, with vimentin expression in the cytoplasm (Figure 6). Immunohistochemistry analysis demonstrated positive vimentin expression in cancer cells, while actin, CK, SMA, desmin, S100, and CD34 were negative. The Ki-67 proliferation index was 10%, leading to a diagnosis of LGFMS. Radiotherapy was initiated 4 weeks postsurgery. At the 6-month follow-up, CT scan revealed disruption of the left MS wall, ethmoid bone, and inferior orbital wall, along with the presence of soft-tissue shadow within the MS (Figure 7).
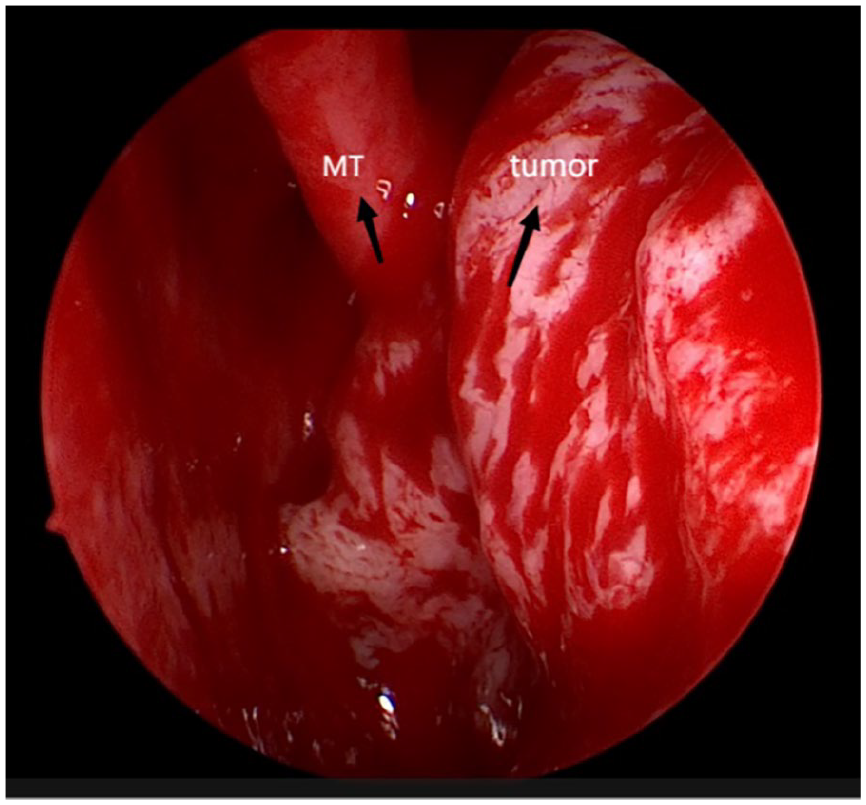
Visualization of a soft-tissue mass within the left middle nasal canal.
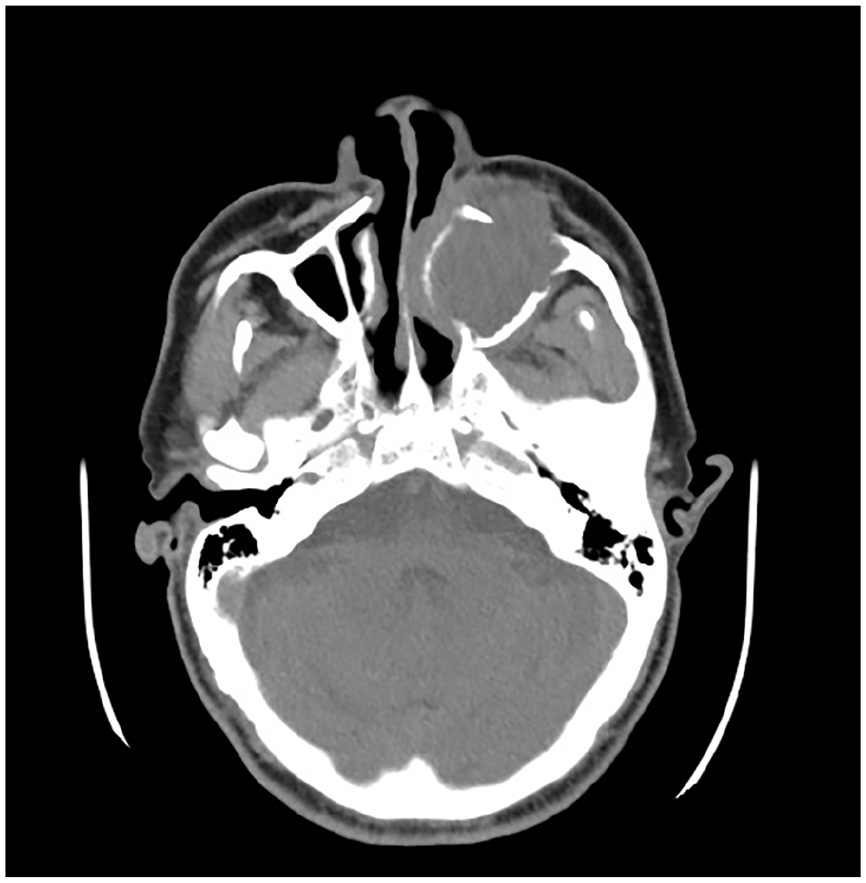
Second preoperative axial CT imaging.
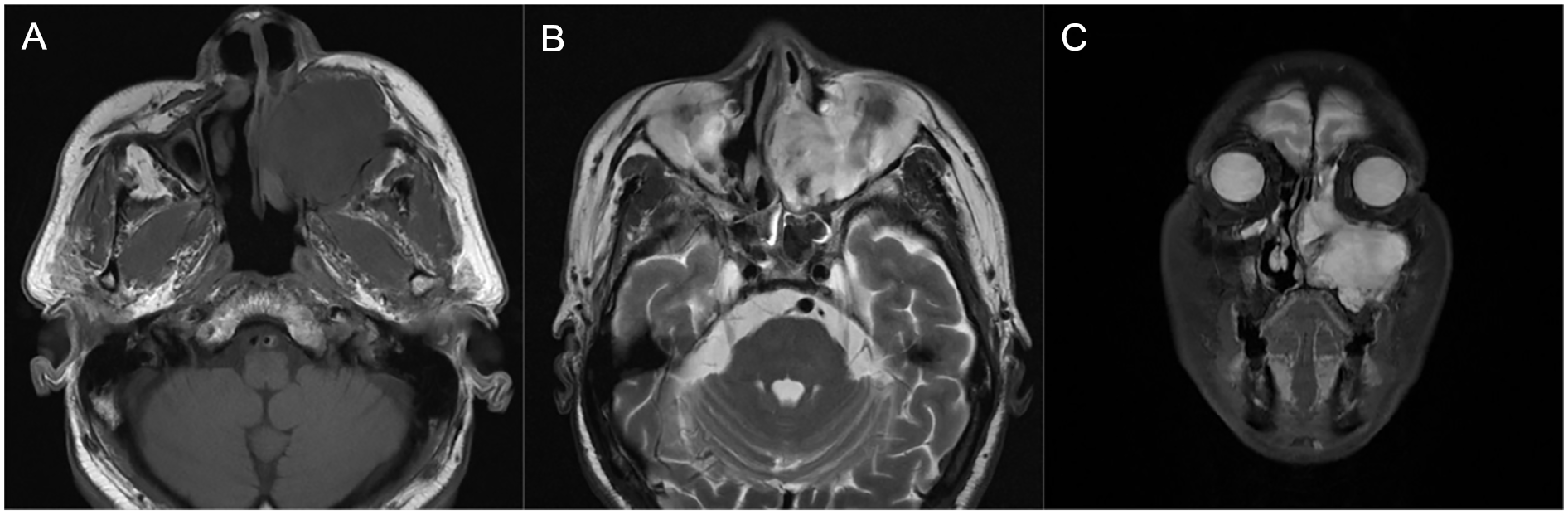
Second preoperative MRI scans displaying the tumor mass within the left MS. (A) TI-weighted images. (B, C) Both T2-weighted images. MS, maxillary sinus.
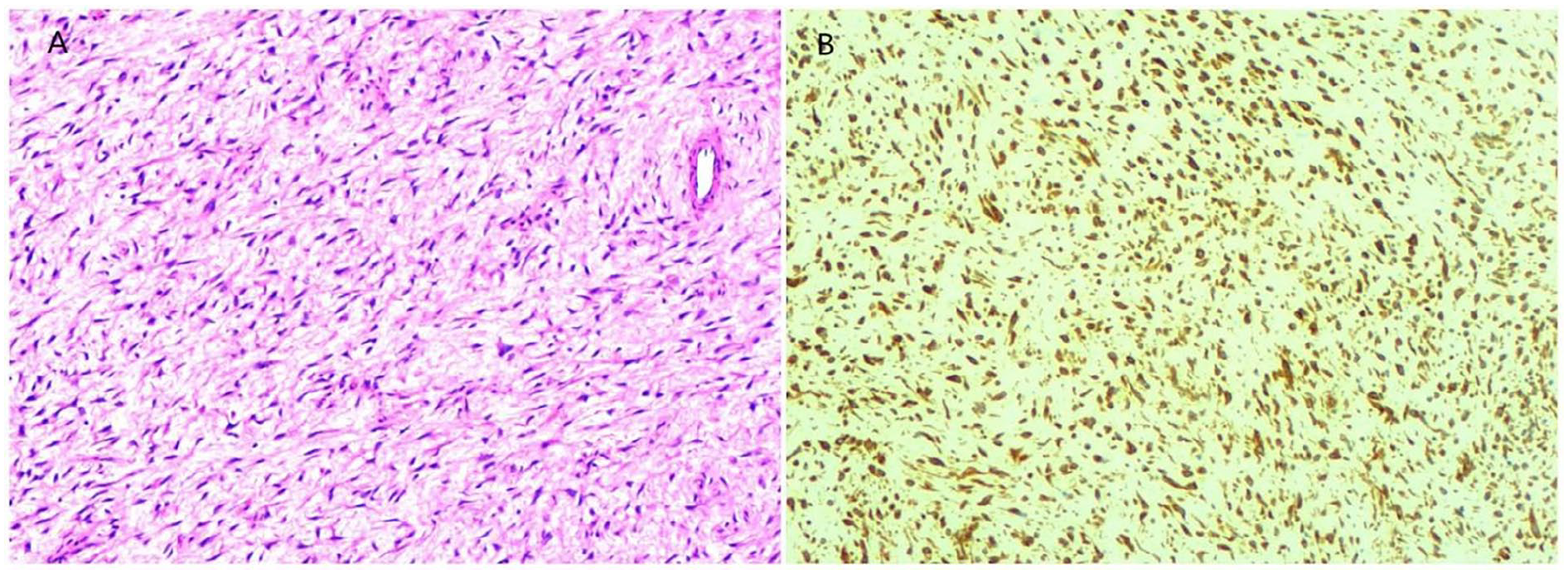
(A) Microscopic examination revealing tumor cells with short fusiform or oval shapes, exhibiting atypia, enlarged nuclei, angular irregular morphology, mitotic images, and extensive mucous degeneration of the interstitium (H&E, magnification ×100). (B) Positive cytoplasmic expression of vimentin in tumor cells (EnVision, magnification ×100).
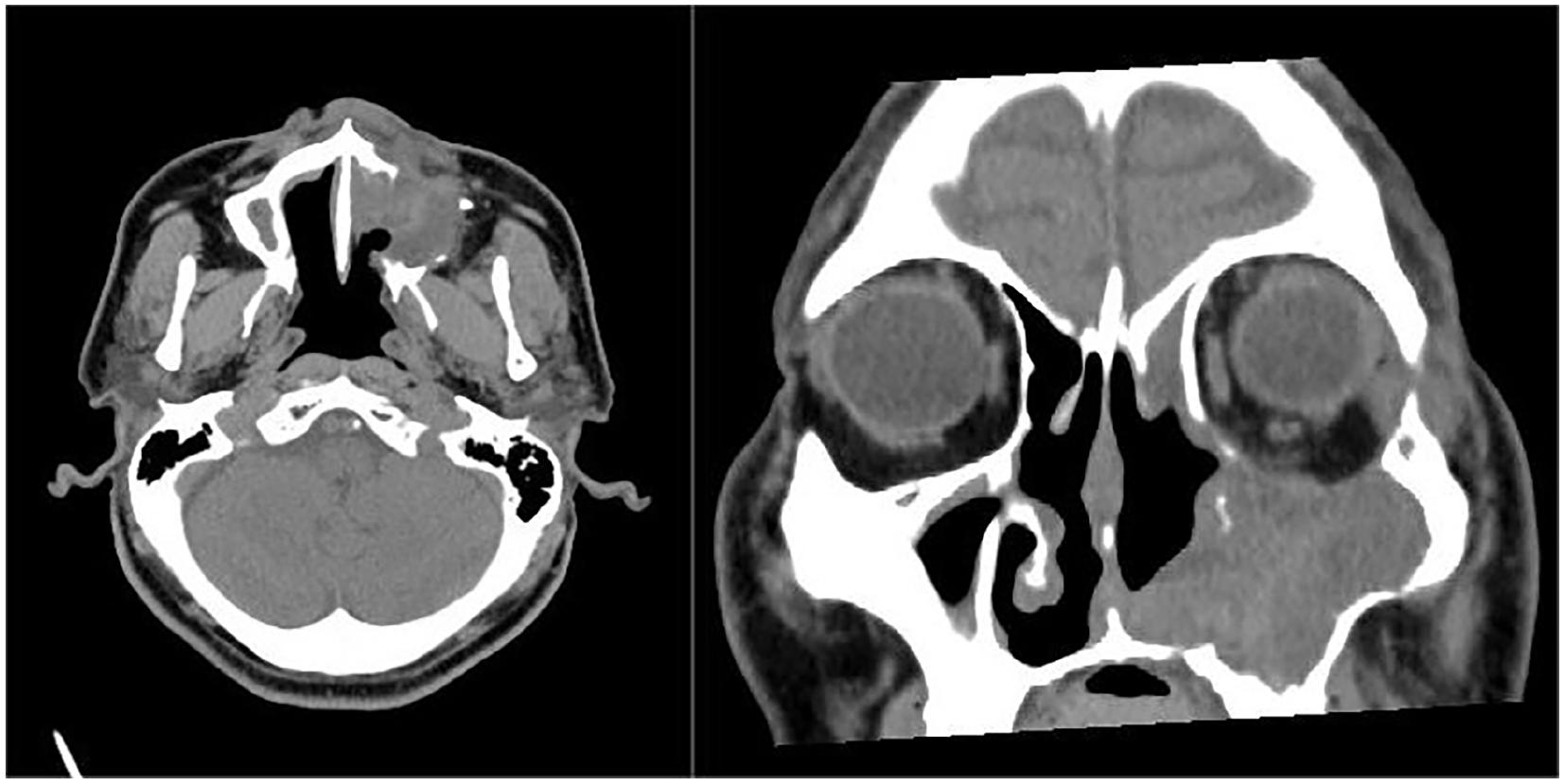
Axial and coronal plane postoperative sixth-month follow-up CT imaging.
The patient underwent a third operation at a prestigious hospital, where a Weber-Ferguson incision was performed to excise the left maxilla and tumor, along with the left infraorbital mass. Immunohistochemistry analysis revealed that AHNAK2, CD34, and CD10 exhibited positive results, whereas S100, CK, desmin, EMA, SMA, MUC-4, and keratin showed negative results. The Ki-67 proliferation index measured at 60%. Despite negative results for FUS, MDM2, and EWSR1 gene rearrangement in the fluorescent in situ hybridization test, the FUS ratio was noted to be 8%. Consequently, the diagnosis was classified as mesenchymal malignancy (FNCLCC II). Eight months following this surgical intervention, the tumor recurred in the left orbital area and is presently being treated with oral anlotinib hydrochloride capsules, a targeted drug for the treatment of advanced soft-tissue sarcoma. 6
Discussion
LGFMS is an uncommon tumor comprised of fibroblastic and myofibroblastic elements, typically found in the deeper layers of the soft tissues of the abdomen, trunk, and proximal extremities. While occurrences in other primary sites such as head, neck, kidney, mesentery, intracranial region, and retroperitoneum are rare, localization in the MS is even more uncommon. 2,7 -10 The age range for LGFMS onset spans from 2 months to 84 years old, with a predilection for young and middle-aged adults, though cases also occur in children and older adults. 5,8
Clinical presentations of LGFMS often manifest as slow-developing, painless solitary masses characterized by a firm texture, acting as a benign tumor. However, LGFMS can display malignant characteristics such as distant metastasis and local recurrence. 11 On CT scan, LGFMS typically presents as an irregular, moderately low-density or isodense mass that erodes adjacent bone with an indistinct boundary with adjacent tissues. Enhanced imaging demonstrates a subtle improvement of the lesions. LGFMS appears hypointense on T1-weighted images and demonstrates mixed signals of hyperintensity and hypointensity on T2-weighted images in MRI, due to its composition of mixed myxoid and fibrous components. 12
The conclusive diagnosis of LGFMS primarily relies on pathological examination. Histomorphologically, the tumor exhibits collagenous hypocellular regions with abundant myxoid nodules. The cancer cells typically display a bland, spindled morphology, occasionally appearing plump, and develop in brief fascicles or a whorl-like arrangement. 13 Importantly, mitotic activity is rare, representing a key characteristic feature of the tumor. 11 Immunohistochemically, tumor cells typically express MUC4, BCL-2, CD99, and vimentin to varying degrees, while they show low expression of CD34, S-100, smooth muscle actin, and desmin. 13,14 Genetically, most of LGFMS cases harbor a fusion gene involving FUS-CREB3L2 as a result of a recurrent translocation (7;16)(q34;p11). 15 LGFMS is a distinct low-grade malignant tumor, and while numerous case reports and series exist in the literature, its diagnosis remains challenging for surgeons. In this case, definitive diagnosis was only achieved after the second postoperative pathology was reviewed at a specialized hospital. Diagnosing LGFMS is particularly challenging for hospitals without access to molecular, genetic, and immunohistochemical laboratories.
The primary treatment for LGFMS typically involves comprehensive surgical removal, often supplemented with radiotherapy and chemotherapy if deemed appropriate, though this approach remains contentious. Even with a decision for radical surgery, there is no guarantee of improving the patient’s condition or altering the course of the disease. Patients frequently encounter frequent and short-term recurrences, along with distant metastases, necessitating multiple surgical interventions. 2,5,16,17 In a study by Evans et al, the long-term follow-up results of LGFMS revealed the recurrence, metastasis and fatality rates of 63.64%, 45.45%, and 42.42%, respectively. 2 In our case, the patient underwent 3 surgeries over the course of a year and relapsed 8 months after the third surgery.
Conclusion
LGFMS is an uncommon soft-tissue tumor with a deceptively bland histological feature and highly malignant characteristics. It is commonly found in the deeper layer of the soft tissues of the trunk or proximal extremity of young adults. Clinical presentations and associated examinations lack specificity, rendering the clinical diagnosis of LGFMS particularly difficult. The conclusive diagnosis primarily relies on pathological assessment and should include genetic and immunohistochemical evaluation. Treatment typically involves excessive resection combined with chemotherapy and radiotherapy if deemed necessary, although the efficacy of these treatments remains controversial. However, strict long-term follow-up is important postsurgery to monitor for possible recurrence or distant metastasis. Given its malignant nature, clinical assessment and therapeutic intervention of LGFMS should be approached with vigilance.
Footnotes
Author Contributions
Dan Zhao: analysis, investigation, writing original draft. Jian Dai: analysis, investigation, review & editing. Yu Hu: analysis, investigation, review & editing. Tao Wang: conceptualizaion, methodology, investigation, analysis, review & editing, supervision.
Data Availability Statement
All relevant data are within the manuscript and its additional files.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Statement/Confirmation of Patient’s Permission
Not applicable.