
Abstract
Patients with chronic rhinosinusitis (CRS) that is refractory to maximal medical and surgical therapy should be evaluated for other primary conditions. Cystic fibrosis (CF), primary immunodeficiency (PID), and primary ciliary dyskinesia (PCD) are potential risk factors for refractory CRS. These conditions present with variable disease severity and diagnosis may be delayed into adulthood. We report a case of a mother-daughter pair with CRS refractory to maximal medical management. The patients were further evaluated and found to have features consistent with CF, PID, and PCD. All 3 are rare disorders and thought to cause CRS in isolation. Patients with refractory CRS should be further evaluated to identify alternative diagnoses and ensure proper management. Refractory CRS may be multifactorial, with different risk factors simultaneously contributing to its persistence.
Background
Chronic rhinosinusitis (CRS) is a prevalent inflammatory disorder of the nasal passages and sinuses. CRS is diagnosed based on 12 weeks or longer of symptoms including nasal pressure, obstruction, discharge, and decreased smell, and confirmed by endoscopic appearance of mucosa and characteristic changes on computed tomography (CT) imaging. 1 Initial treatment for CRS includes systemic antibiotics, intranasal corticosteroids, short courses of systemic steroids, and topical steroid irrigations. In cases where medical therapy is insufficient, patients undergo endoscopic sinus surgery (ESS).2,3 Although this approach is successful in most cases, a subset of patients continues having inflammation and sinus symptoms after ESS despite being on appropriate medical therapy.
Refractory cases of CRS are challenging to manage. These patients should be further evaluated to rule out secondary CRS. In this case report, we present a mother-daughter duo with refractory CRS with features consistent with cystic fibrosis (CF), primary immunodeficiencies (PID), and primary ciliary dyskinesia (PCD). All 3 are rare disorders and classically thought to cause CRS in isolation.
Case Details
Clinical Presentation and History
A mother (age 58) and daughter (age 20) presented with chronic bilateral nasal blockage associated with mucopurulent discharge, postnasal drip, anosmia, and facial pain. Their symptoms were unresponsive to frequent antibiotic courses. They had been managing their symptoms with saline rinses and steroid nasal sprays. Neither patient endorsed cough nor chest pain. Past medical history was unremarkable. Allergy testing was negative for both. The mother was a lifelong nonsmoker while the daughter previously smoked for a few years, averaging less than 5 cigarettes per week.
Physical Examination and Preoperative Investigations
Endoscopy revealed bilateral mucopurulent discharge in both patients and bilateral grade 1 polyps in the mother only. Sinus CT scan showed bilateral patchy opacification of the paranasal sinuses with ostiomeatal complex obstruction, in addition to maxillary sinus hypoplasia (Figure 1). The Lund-Mackay scores were 16/24 and 12/24 in the mother and daughter, respectively. Chest X-ray, CT, and spirometry were all within normal limits.
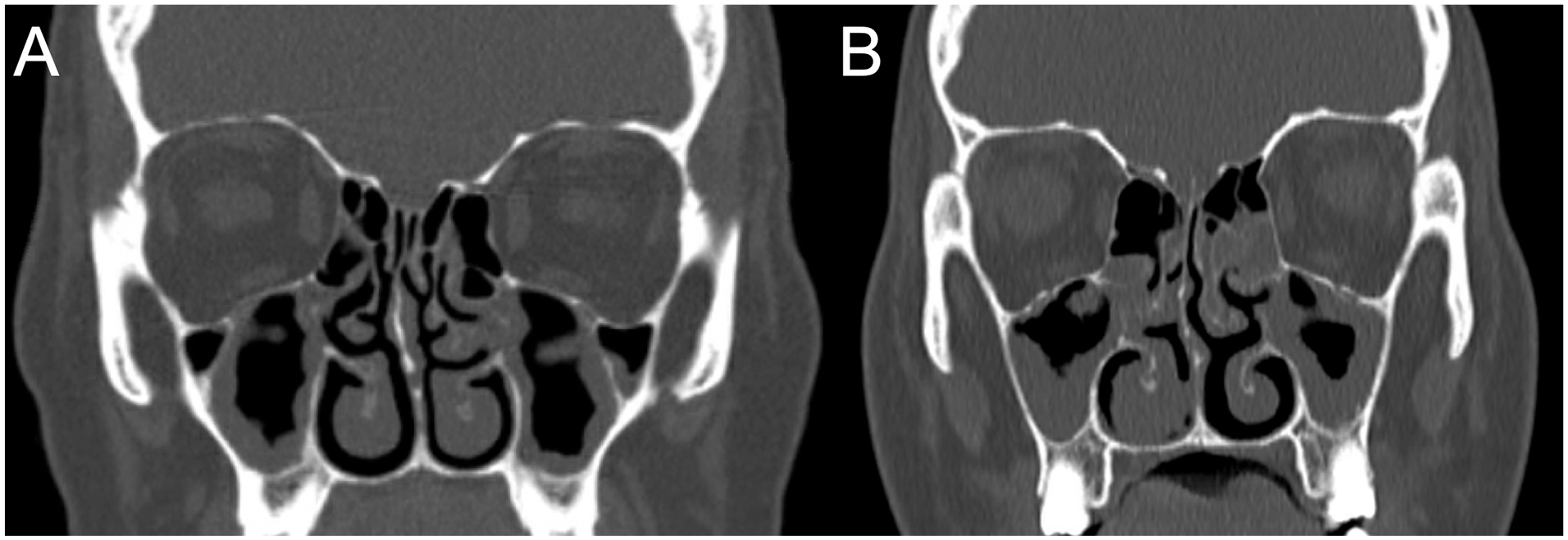
Sinus CT scans of the (A) mother and (B) daughter, showing bilateral patchy opacification of the paranasal sinuses with ostiomeatal complex obstruction. Maxillary hypoplasia is notable: normally, the maxillary sinus floor is below the nasal floor. CT, computed tomography.
Diagnosis and Management
The mother was diagnosed with CRS with nasal polyps and the daughter had CRS without nasal polyps. Daily budesonide rinses were initiated in both patients but neither responded well; both received ESS a few years later. While short-term outcomes were favorable, their CRS symptoms returned soon after in both patients.
Postoperative Investigations
Both patients were evaluated for CF. The sweat chloride levels were consistently elevated at 61 mmol/L in the mother and 53 mmol/L in the daughter (normal <40 mmol/L). However, genetic testing for disease-causing mutations in the CF transmembrane regulator (CFTR) gene was negative for both patients. A few years later, the mother also developed borderline pancreatic insufficiency, with fecal elastase 190 µg/g (normal >200 µg/g).
The mother was also investigated for PID. Quantitative immunoglobulin (Ig) levels were IgG 7.6 g/L (normal 7-16 g/L), IgA 0.03 g/L (normal 0.70-4.00 g/L), and IgM 0.35 g/L (normal 0.40-2.30 g/L). These values were repeated and remained stable 1 year later. In response to the pneumococcal polysaccharide vaccine, her antibody levels were under 0.3 mcg/mL for all serotypes (normal >1.3 mcg/mL). Flow cytometry revealed elevated T-cell populations: CD3 was 0.85 (normal 0.62-0.84) and CD8 was 0.36 cells/100 cells (normal 0.12-0.32). B-cells were within normal limits: CD19 was 0.10 cells/100 cells (normal 0.05-0.23).
To query PCD, the daughter consented to a mucus membrane biopsy. Electron microscopy revealed short cilia ranging from 2 to 3 μm (normal 5-7 μm) and decreased number of cilia per cell (Figure 2). The ultrastructure was normal: mean outer dynein arm count was 8.8 (normal range 8-9) and mean inner dynein arm count was 7.6 (normal range 4-9; Figure 3). There was normal 9 + 2 microtubular symmetry with nexin links and radial spokes. Testing for mutations in known PCD genes revealed a heterozygous substitution at position 2226 of DNAH11 (c.2226A>G, p.(Ile742Met)).
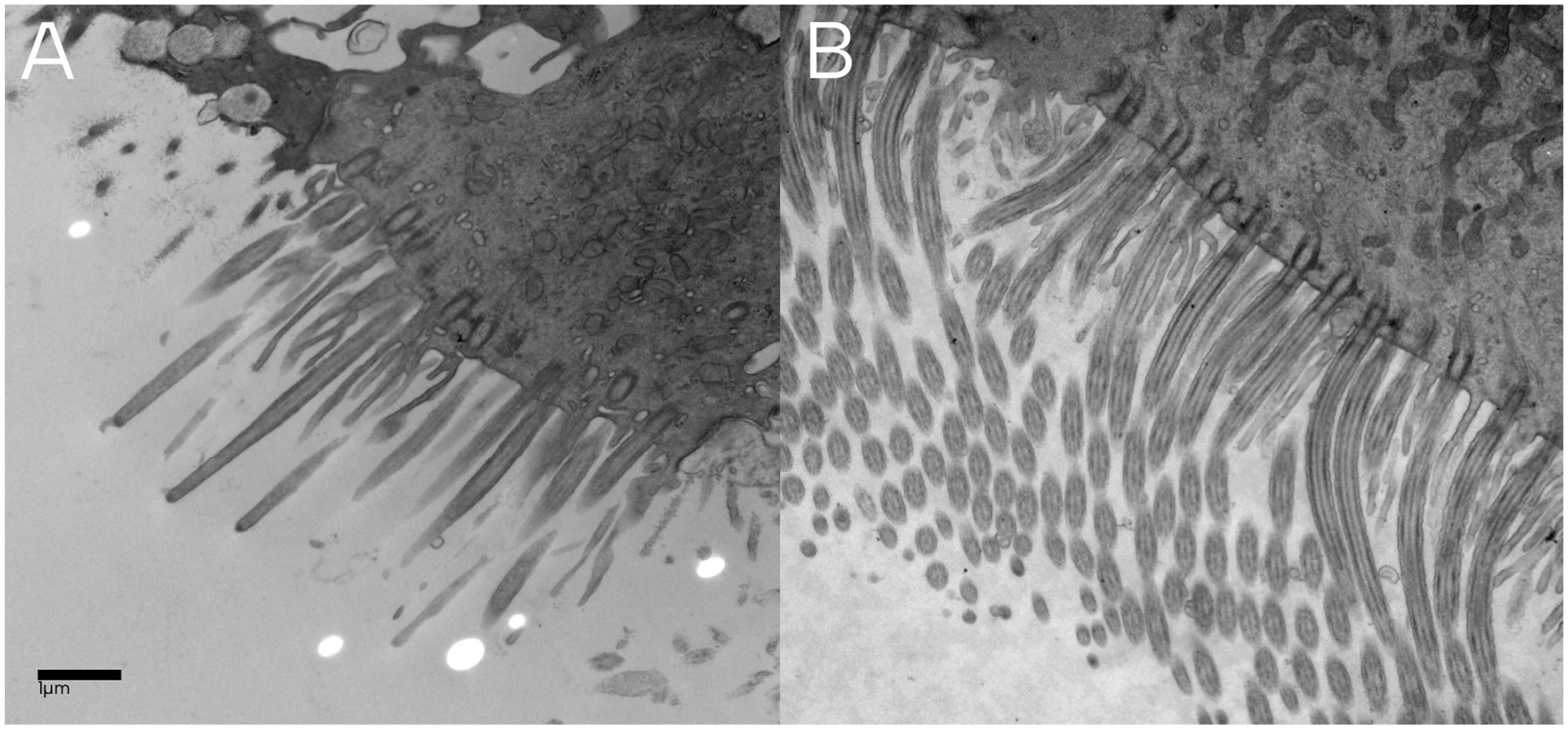
(A) Shortened (2-3 μm) and reduced number of cilia in the daughter. (B) Normal cilia length and number in a healthy patient.
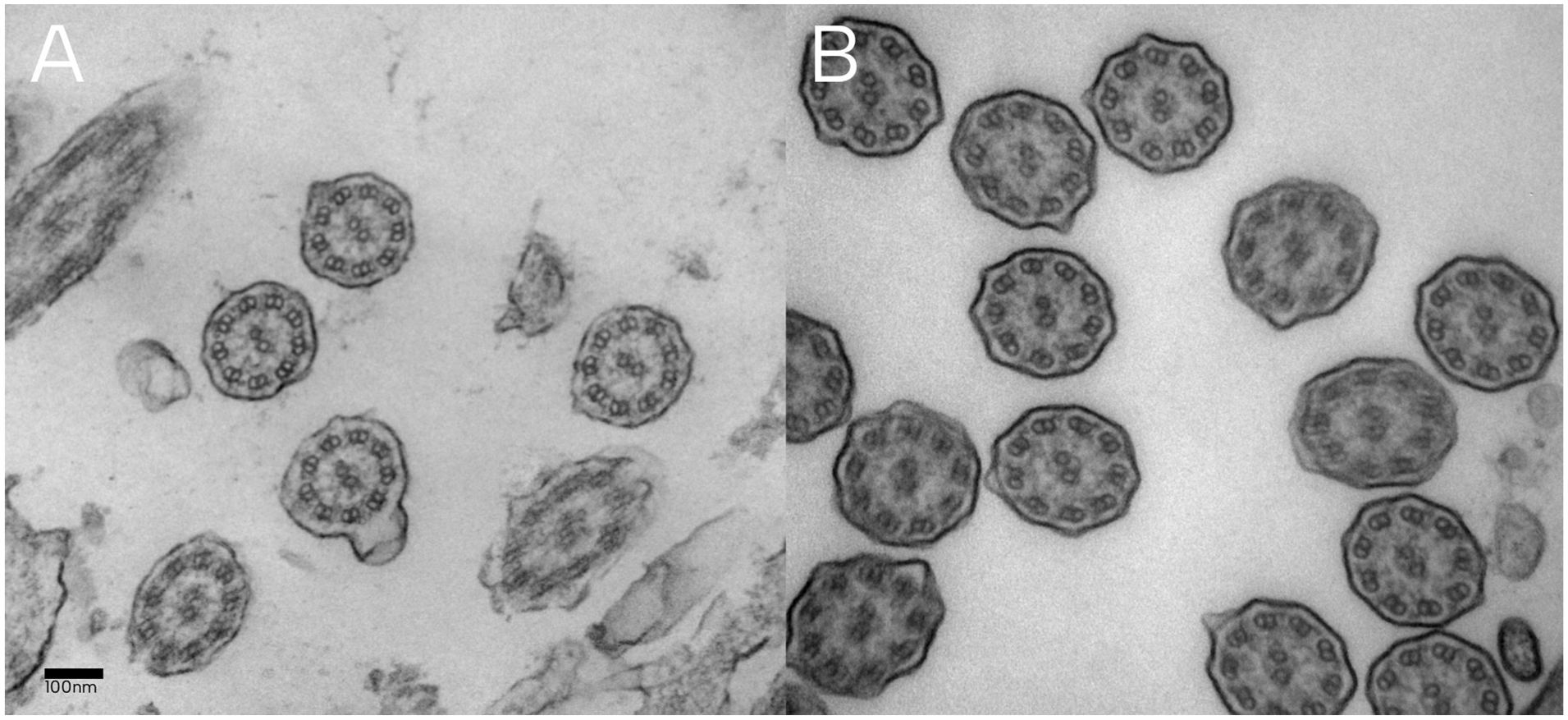
(A) Ciliary cross sections in the daughter showing normal ultrastructure. (B) Cross sections of cilia in a healthy patient.
Discussion
Despite maximal medical and surgical therapy, select patients have persisting CRS symptoms. In these cases, secondary CRS should be considered. The mother-daughter pair described above with recalcitrant CRS had features consistent with atypical CF, PID as well as PCD.
Atypical CF
CF is characterized by mucostasis due to inherited mutations in CFTR, leading to chronic sinopulmonary infection and malabsorption. In classic CF, these symptoms manifest in early childhood. Diagnostic criteria for classic CF include laboratory evidence of CFTR dysfunction (sweat chloride >60 mEq/L), identification of 2 disease-causing CFTR mutations, and CF phenotype in at least one organ system.4,5
Atypical CF is a milder form which does not strictly meet these criteria. Sweat chloride may be borderline, ranging between normal (<40 mEq/L) and pathologic values (>60 mEq/L).5-7 Disease-causing mutations, when identified, are frequently singular.6,7 Dysfunction is often confined to one organ system with milder symptoms than the classic form. As a result, the diagnosis of atypical CF is often made in late adolescence or adulthood. A recent meta-analysis showed a significantly higher prevalence of CFTR-mutation carriers in CRS patients compared to the general population. 8 This suggests that atypical CF should be queried in patients with recalcitrant CRS.
In our case, both the mother and daughter presented with features consistent with atypical CF. Sweat chloride levels fell in the intermediate-high range, suggesting CFTR dysfunction despite the absence of identifiable disease-causing mutations. The presence of CRS, in addition to constipation and laboratory-confirmed pancreatic insufficiency in the mother, are phenotypes consistent with CFTR dysfunction. Furthermore, CT in both patients revealed maxillary hypoplasia, which is characteristic of CF (Figure 1). 9
Primary Immunodeficiency
PIDs are variable in cause and phenotype. While antibody deficiencies are most common, any defect in the components of innate and adaptive immunity may lead to symptoms. Recalcitrant CRS is the most common clinical manifestation: it has been reported that up to 50% of patients with recalcitrant CRS have an immunodeficiency.10,11 Given this high prevalence, all patients with recalcitrant CRS should be worked up for an immunodeficiency. History of recurrent sinopulmonary and gastrointestinal infections, in addition to prior positive cultures of encapsulated or opportunistic organisms, should raise strong suspicion for a PID. 12 Laboratory workup including complete blood count with differential and serum immunoglobulin levels should be performed. Although unreliable, antibody response to vaccines and flow cytometry to enumerate B- and T-cells may be considered. 12
Common variable immunodeficiency disorder (CVID) is the most frequent symptomatic PID in adults. 13 Antibody failure is classically late onset. Most cases are polygenic, and penetrance and expressivity of the disease varies widely. The International Consensus Document on CVID proposed the following diagnostic criteria in 2015: (1) at least one clinical manifestation of immune dysfunction, (2) the IgG level must be repeatedly low in at least 2 measurements 3 weeks apart, (3) IgA and IgM must be low, and (4) other causes of hypogammaglobulinemia must be excluded. 14 Although not required for diagnosis, genetic studies and antibody response to vaccines were recommended where possible. However, the best parameters to diagnose CVID remain controversial.12,13 Treatment for CRS specifically in the context of PID includes immunoglobulin replacement, although its effectiveness for CRS is met with mixed results.11,15
In our case, the mother had persistently low IgG, IgA, and IgM levels in measurements taken 1 year apart, suggesting CVID. In further support of this, antipneumococcal antibody levels were low in response to both the 13- and 23-valent polysaccharide vaccines. Flow cytometry profiles widely vary in CVID; our patient had normal peripheral B-cells but higher-than-normal values in select T-cell populations, which is consistent with previous reports.16,17
Primary Ciliary Dyskinesia
In PCD, structural defects result in impaired mucociliary clearance leading to chronic sinopulmonary infection and otitis media. Dynein arm abnormalities are the most common defects, leading to dyssynchronous ciliary beating. 18 Due to the role of cilia in establishing left-right asymmetry in embryogenesis, approximately 50% of patients have associated situs inversus. 18
Delayed diagnosis is common in PCD, likely due to variability in severity and lack of diagnostic standards. 19 Recent guidelines suggest that 2 of the following clinical features should be present for PCD to be suspected: unexplained respiratory distress during infancy, year-round daily coughing or evidence of bronchiectasis on CT, year-round daily nasal congestion or pansinusitis, and organ laterality defect. 19 Further workup should include genetic testing and nasal nitric oxide measurements where accessible. To date, 50 disease-causing mutations have been identified in PCD and the inheritance pattern is autosomal recessive in most patients. 20 In patients where a single or no pathogenic variant is identified, electron microscopy of the cilia is recommended to evaluate for structural defects. 19
In our case, genetic testing in the daughter revealed one mutation in DNAH11. Mutations in this gene are a common cause of PCD without ciliary ultrastructural defects. 21 In keeping with this, electron microscopy of her cilia revealed short cilia with normal ultrastructure (Figures 2 and 3).
Conclusion
Patients with refractory CRS should be evaluated for secondary CRS. CF, PID, and PCD present with variable disease severity and diagnosis may be delayed, which impedes appropriate management. These disorders are relatively rare and generally believed to cause CRS in isolation. This case is unique as features of all 3 disorders coexist in a mother-daughter pair. Refractory CRS may be multifactorial, with features of multiple diseases simultaneously contributing.
Footnotes
Availability of Data and Materials
Not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Individual informed patient consent was obtained. Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
The patients have consented for this case report to be published.