
Abstract
Cogan syndrome is an autoimmune disease characterized by vestibular symptoms, bilateral sensorineural hearing loss, and inflammatory ocular manifestations, which may be accompanied by systemic vasculitis. We herein present the case of a patient with bilateral sensorineural hearing loss who presented with pain over her cochlear implantation incision site. She was later found to have evidence of ocular disease and underlying vasculitis leading to a diagnosis of Cogan syndrome.
Introduction
Cogan syndrome is a rare autoimmune vasculitis with approximately 450 cases reported in the literature. It is characterized by intralocular inflammation and vestibulocochlear dysfunction, with systemic vasculitis present in about 10% of cases. 1 This disease was first described in 1934 by Morgan and Baumgartner 2 as non-syphilitic interstitial keratitis in conjunction with vestibulocochlear dysfunction. Eleven years later, in 1945, David Cogan clinically defined this syndrome with 4 additional cases. This disease’s ocular and otologic manifestations can present up to several months apart, making the diagnosis challenging. 3 In addition, due to the rarity of this disease, it requires a high level of clinical suspicion, and, likely, many are incorrectly diagnosed as idiopathic progressive hearing loss, autoimmune inner ear disease, as well as idiopathic recurring keratitis. 1
Case Report
This is the case of a 44-year-old female patient with bilateral cochlear implants initially placed following a rapid progressive hearing loss, with the left implant placed in September 2021 and the right implant in March 2022 at an outside hospital. Her postoperative course was unremarkable, and the patient made a full recovery. In May 2022, the patient noted redness and edema over her left implant site and presented to her surgeon. She was presumed to have an infection that was then treated with cephalexin, trimethoprim-sulfamethoxazole, and amoxicillin-clavulanate sequentially without resolution of her symptoms.
She presented to the emergency department at the senior author’s institution in June 2022 due to pain at her left cochlear implant site. Her physical examination at that time revealed overlying erythema of the right implant site and partial extrusion of the left cochlear implant (Figure 1). She was also found to have multiple areas of large erythematous papular lesions of her forehead, lower extremity, and bilateral upper extremities, which progressed into extensive ulcerations (Figure 2). The patient’s labs at the time of admission revealed a hemoglobin of 10.8 (normal range, 11.5-15.0) and elevated inflammatory markers, including erythrocyte sedimentation rate (ESR) (62, normal range 0-20) and c-reactive protein (CRP) (7.5, normal range 0.0-1.0) without evidence of leukocytosis and normal antineutrophil cytoplasmic antibody (ANCA), myeloperoxidase antibody, and serine protease antibody (PR3). The patient voiced a strong desire to have her implants removed due to her significant pain.
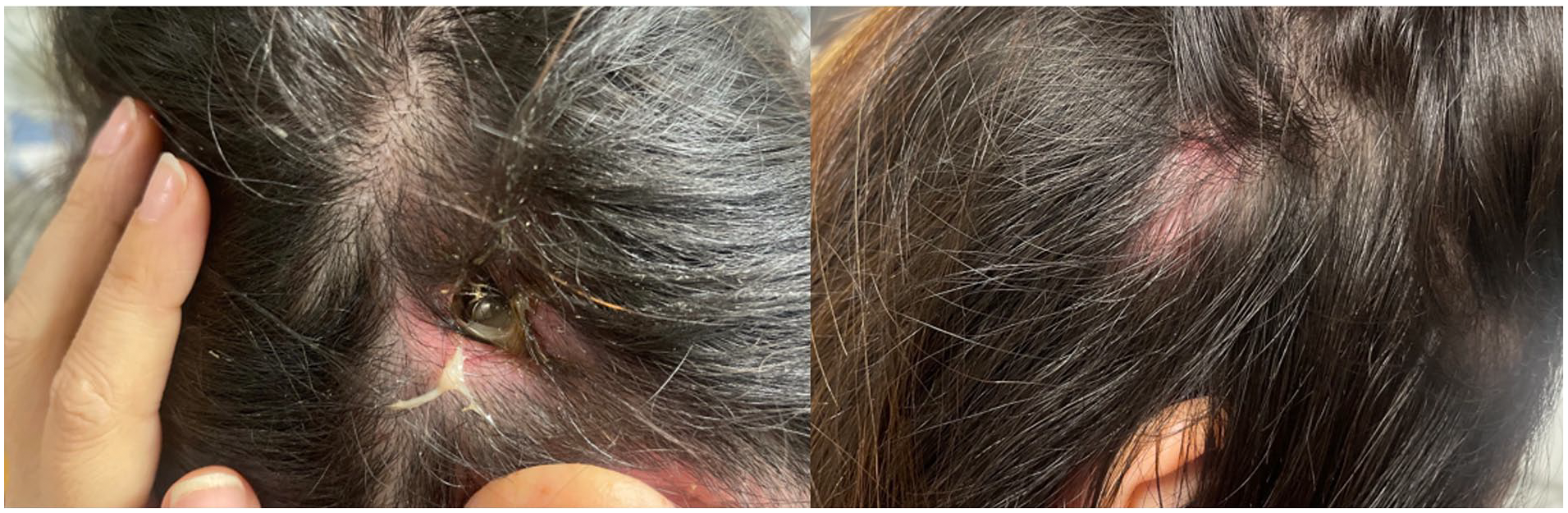
Partial extrusion of (A) left cochlear implant and erythema of the right (B) implant noted during the patient’s initial admission.
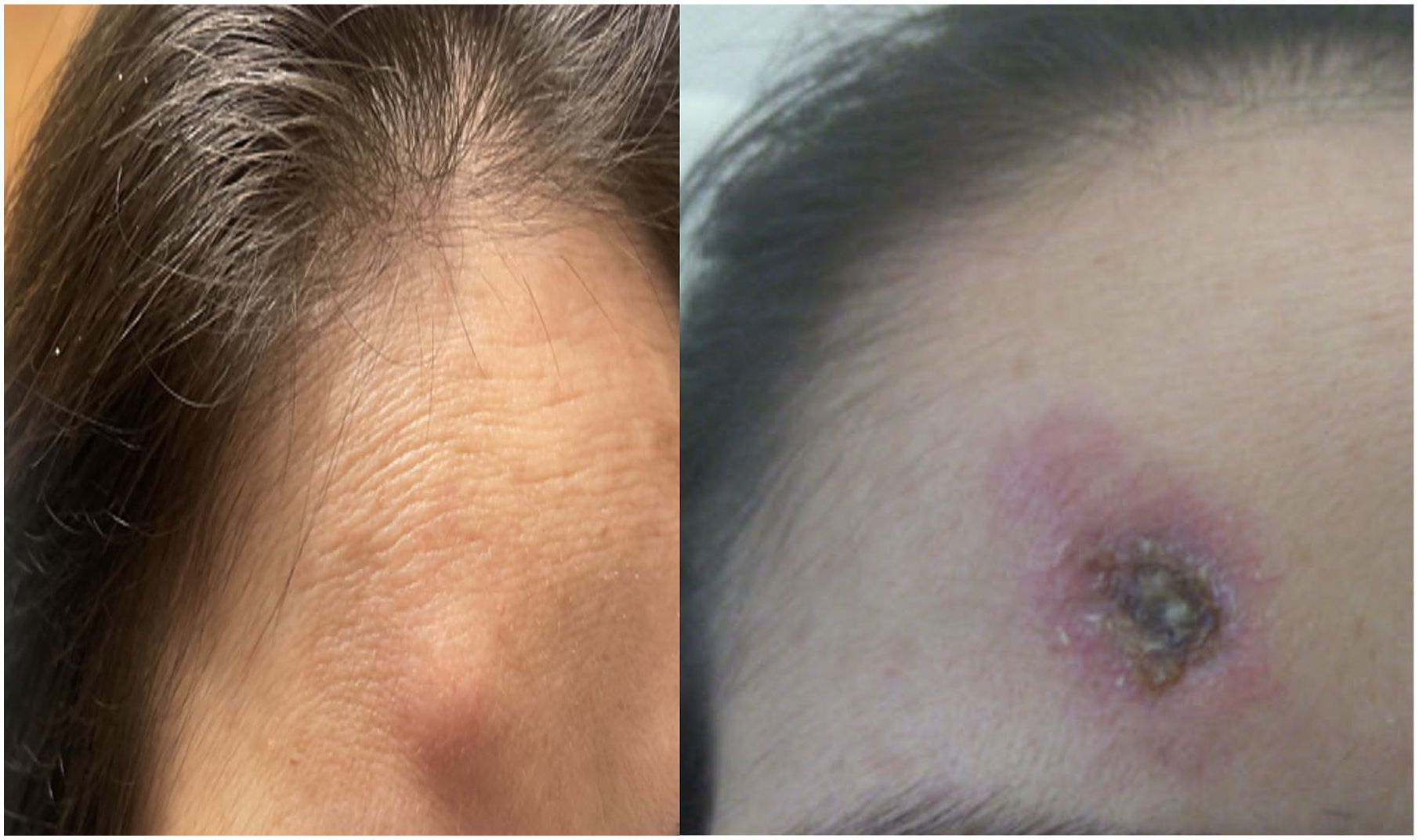
Patient’s photo demonstrating (A) large papular lesion of the forehead, which progressed into a large ulceration (B) in 14 days.
The implants were subsequently surgically removed in July 2022. Approximately 6 days postoperatively, dehiscence was noted at the surgical site (Figure 3). A biopsy of her forehead lesion with Fite staining was read to be positive for atypical tuberculosis (TB). The patient was started on a multidrug regimen for presumed disseminated TB. Two weeks later, the patient developed nodular scleritis of her right eye (Figure 4). During this time, the patient was also found to have progression of her arm lesions with ulcerative lesions also noted on her face bilaterally. A chest computed tomography (CT) was ordered, which was remarkable for circumferential wall thickening of the aortic arch, the distal-most ascending thoracic aorta, the proximal brachiocephalic artery, and the proximal left common carotid artery. Two ulcerations were also noted along the thoracic aorta, with the largest measuring up to approximately 3 mm in depth (Figure 5). In addition, there was also narrowing of the proximal descending thoracic aorta with dissection of the aorta noted. Given the patient’s clinical picture, including bilateral sensorineural hearing loss, scleritis, and evidence of vasculitis with predominant aortic involvement, the patient was thought to have Cogan syndrome. The multidrug therapy for presumed tuberculosis was stopped, and she was subsequently placed on prednisone with marked improvement of her pain and ulcerative lesions of the skin. The patient is now on maintenance therapy with weekly methotrexate and every other week, adalimumab. Reimplantation of the cochlear implants may be considered once the patient’s vasculitis is managed.
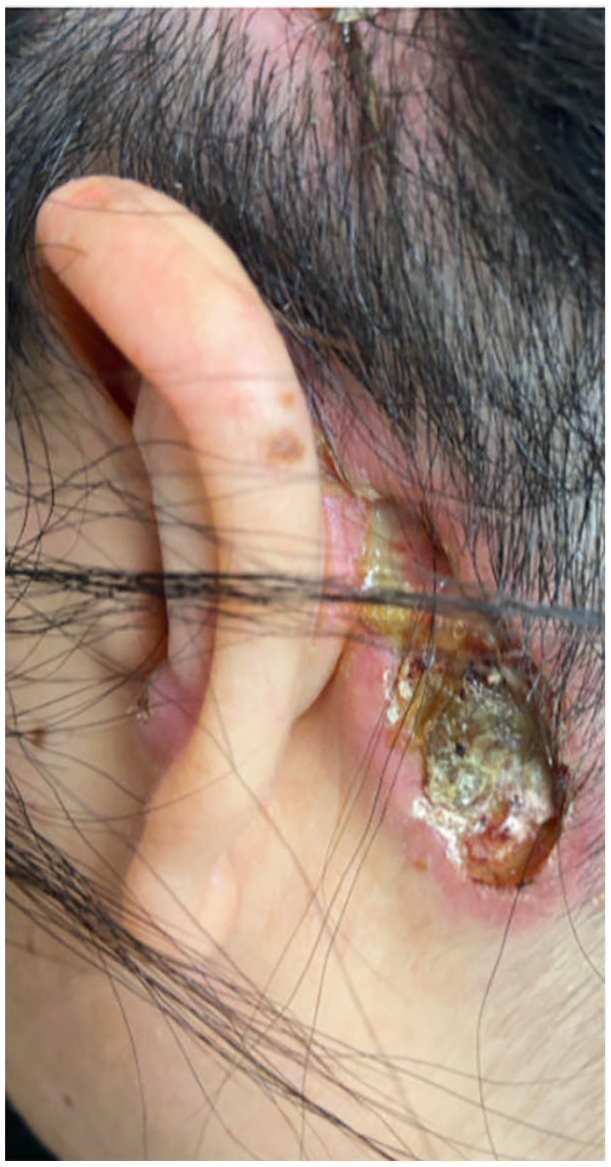
Breakdown of the explantation wound at 14 days.
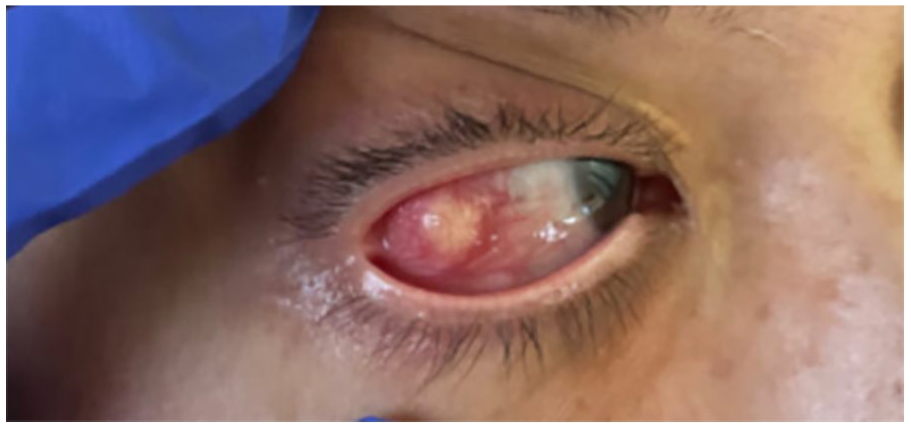
Patient’s photo 2 weeks following bilateral cochlear implant removal showing nodular scleritis of the right eye.
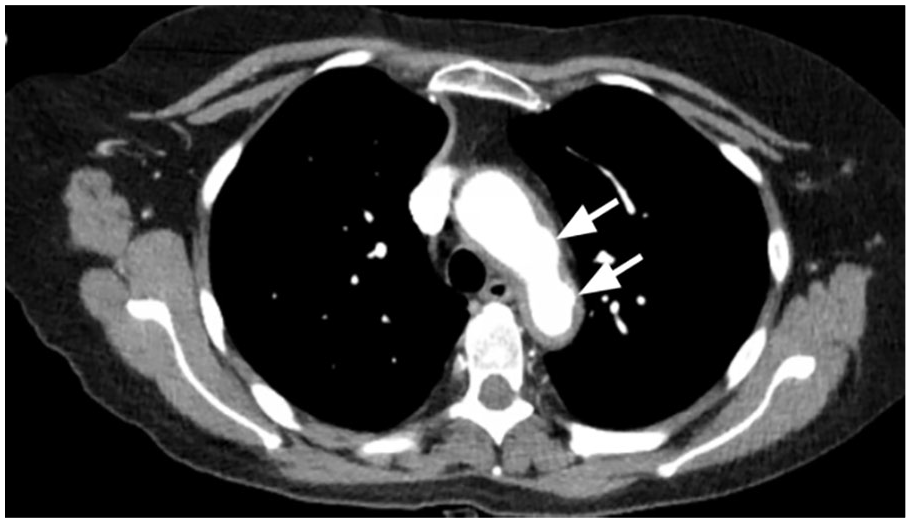
Axial chest CT demonstrating 2 ulcerations noted along the thoracic aorta depicted with arrows. The largest measuring up to approximately 3 mm in depth.
Discussion
Cogan syndrome is a rare disorder encompassing inflammatory eye disease and vestibulocochlear symptoms, of which vasculitis is considered the pathologic mechanism.4,5 Cogan first described this disease in 1945 as a “syndrome of non-syphilitic interstitial keratitis (IK) and vestibuloauditory symptoms” in a report of 4 case studies. 6 In 1980, Haynes et al further characterized the disease as a typical or atypical pattern. Typical is defined as non-syphilitic IK, vestibular symptoms similar to Ménière’s disease, and acute onset sensorineural hearing loss, eventually leading to deafness within 2 years. Atypical Cogan syndrome is classified as vestibulocochlear symptoms associated with an inflammatory eye disease other than IK or when the interval between the onset of vestibulocochlear and ophthalmologic symptoms is greater than 2 years.7,8 Caucasian young adults are most commonly affected with a median age of onset of 29 years. There does not appear to be a gender-specific prevalence or a hereditary component. 5
The vestibulocochlear manifestations of this disease can mimic those of Ménière’s disease, with vertigo, nausea, vomiting, tinnitus, and severe sensorineural hearing loss. The hearing loss is typically bilateral, as is the case in this patient. 9 There are a number of hallmark histopathologic findings which may explain the vestibulocochlear dysfunction, including plasma cell and lymphocyte infiltration of the cochlea and auditory neuronal loss, endolymphatic hydrops, degeneration within the Organ of Corti, new bone formation, and atrophy of the vestibulocochlear nerve. 10
The most common ocular finding in Cogan syndrome is IK but may include other inflammatory ophthalmologic conditions, including scleritis, episcleritis, uveitis, and conjunctivitis, among others. Further examination commonly reveals ciliary injection with evidence of iritis and opacities in the corneal stroma and a nonuniform granular corneal infiltration. 9 The ophthalmic disease course generally waxes and wanes with periods of remission. Blindness is a rare consequence. 9 Studies have shown that cardiovascular, neurological, and gastrointestinal systemic manifestations are also present in a minority of cases.5,9 Cardiovascular manifestations typically involve the aorta, as was the case in this patient. 11
Cogan syndrome is a challenging diagnosis due to its gradual onset of symptoms and lack of confirmatory lab testing. While there is no proven therapy, the diagnosis is typically confirmed following favorable response to corticosteroids, as was seen with this patient. Treatment with tumor necrosis factor alpha (TNF-α) blockers resulted in sustained clinical remission in some subjects. 12 Cochlear implantation has also been performed in this population as the prognosis for hearing loss is typically poor and irreversible . 13
This is a unique case involving a patient who initially presented with complaints related to her cochlear implants and was later diagnosed as having Cogan’s syndrome. There was initial concern for infection by the outside implant surgeon in this patient, but the lack of response to antibiotics ruled out a typical bacterial infection. In addition, the cultures taken during admission were notably negative, and there was no leukocytosis on lab work. Although the patient was initially presumed to have a tuberculosis infection based on the Fite stain, further evaluation of the Fite staining showed a single possible positive site. A mycobacterial infection was ruled out due to the lack of improvement in the patient’s clinical course. The new scleral nodule and the CT findings of large vessel vasculitis, vestibulocochlear dysfunction, ocular inflammation, and cutaneous manifestations were collectively highly suggestive of a systemic inflammatory process. Wegener’s granulomatosis with polyangiitis, Behçet, and Vogt–Koyanagi–Harada were considered, but ultimately Cogan syndrome seemed most likely given that her vasculitis predominantly involved large vessels.
Cogan syndrome is unique in that it causes large vessel (eg, aorta) vasculitis, inflammatory eye disease, and hearing loss. Another form of vasculitis, granulomatosis with polyangiitis, was considered but ruled out with multiple negative ANCA tests and a negative biopsy from the soft tissue. GPA causes vasculitis in smaller blood vessels and does not typically cause ocular surface inflammation similar to Cogan syndrome. Vogt–Koyanagi–Harada disease was ruled out based on a lack of vision loss, hair/skin color pigment loss, and meningeal involvement. In patients with VKH, the target for the autoimmune process is melanocytes. We considered and ruled out Behçet’s disease based on a lack of oral or genital lesion. Finally, we also considered mycobacterial infection but that was ruled out with multiple quantiferon gold tests and a negative culture from a lesion.
Conclusion
We present the case of a patient with a history of severe bilateral hearing loss who presented due to pain over her cochlear implant incision and was found to have Cogan syndrome. The ocular and vestibulocochlear systems are most commonly affected, with many patients experiencing negative systemic sequelae related to the underlying vasculitis. This is often a challenging diagnosis requiring a high index of suspicion, as early initiation of therapy may help reduce long-term morbidity.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Beyond Tinnitus App., xTinnitus, NeuroMed Care LLC, Cactus Medical LLC., Elinava Technologies, NXT Biomedical, Massimo.
Submission Statement
This article is original and has not been submitted elsewhere in part or in whole.