
Abstract
Neuromyelitis optica spectrum disorder (NMOSD) is an uncommon antibody-mediated disease of the central nervous system. Its classic presentation includes long segments of spinal cord inflammation, optic neuritis with or without intractable vomiting, and hiccups. Here, we described a case of a 39-year-old woman with an atypical presentation of vertigo, which was finally diagnosed as NMOSD by a positive serum aquaporin-4 antibody.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune demyelinating disorder, first described by Eugène Devic and Fernand Gault in 1894. 1 As the targets of autoantibodies, the aquaporin-4 water channel was first identified in 2014 2 and then the myelin oligodendrocyte glycoprotein in 2012. 3 Its core clinical characteristics include optic neuritis (subacute vision loss), acute myelitis (characterized by motor, sensory, or autonomic deficits), area postrema syndrome (intractable nausea, vomiting, and/or hiccups), and brainstem syndrome (diplopia and cranial nerve palsy). 4 The first-line treatment for an acute attack was high-dose corticosteroids (1 g of methylprednisolone daily for 5 days and then taper) and plasma change. 5 Because early treatment is of the utmost importance to reduce neurological sequelae, we must know the variable presentation of NMOSD to make a timely diagnosis.
Here, we report a case of NMOSD with an unusual initial presentation, rapid progression, and reasonable response to high-dose steroids.
Case report
A case of a 39-year-old female visited the emergency room with the initial presentation of dizziness for 4 days. Her dizziness was related to head motion, and each episode could last for 30 minutes. She could not perform the Romberg test, but there was no dysmetria in the finger-to-nose test. No obvious gaze nystagmus was noted, but her dizziness became worse when she turned her head to the right during the Dix–Hallpike test and supine roll test. Therefore, she received the Epley maneuver at first and antivertigenous drugs after discharge. Two days later, she revisited our otolaryngology clinic because of progressive dizziness and new-onset hiccups. Her lab data showed her WBC count 21100/μL with 83% neutrophils, low procalcitonin (< .05), and elevated CRP (12.46 mg/dL). Because of her poorly controlled dizziness and progressive imbalance, she was admitted for intravenous diazepam and chlorpromazine on the seventh day. However, she developed direction-changing gaze nystagmus, progressive hiccups, and mild dysphagia and dysarthria the next day. She could not even move or protrude her tongue. Then, we consulted the neurologist and arranged for a brain MRI (Figure 1), which showed abnormalities over the left dorsal medulla. Autoantibodies associated with connective disorders (C4, rheumatic factor, IgG, antiDNA, antithrombin 3 antibodies, antibeta 2 GPI antibodies, and ENA) were negative, except for elevated C3. Lumbar puncture showed clear cerebrospinal fluid (CSF) with predominant lymphocytes pleocytosis, normal protein levels, a normal glucose CSF–plasma ratio, and normal LDH. Viral encephalitis with brainstem involvement could not be ruled out; therefore, we checked CSF viral PCR (enterovirus, HSV, VZV, CMV, EBV, and adenovirus), TB PCR, bacterial antigens (S. pneumonia, H. influenza, N. meningitis, and Cryptococcus), and cultures, all of which were negative. Finally, on the 17th day, the data revealed a positive aquaporin-4 antibody (AQP4-Ab) indicating NMOSD. Magnetic resonance imaging showing the dorsal medulla lesion. (A) (T2)-hyperintensity, (B) (T1)-hypointensity, (C) (T1 with contrast)-no enhancement, and (D) (Diffusion-weighted image)-hyperintensity.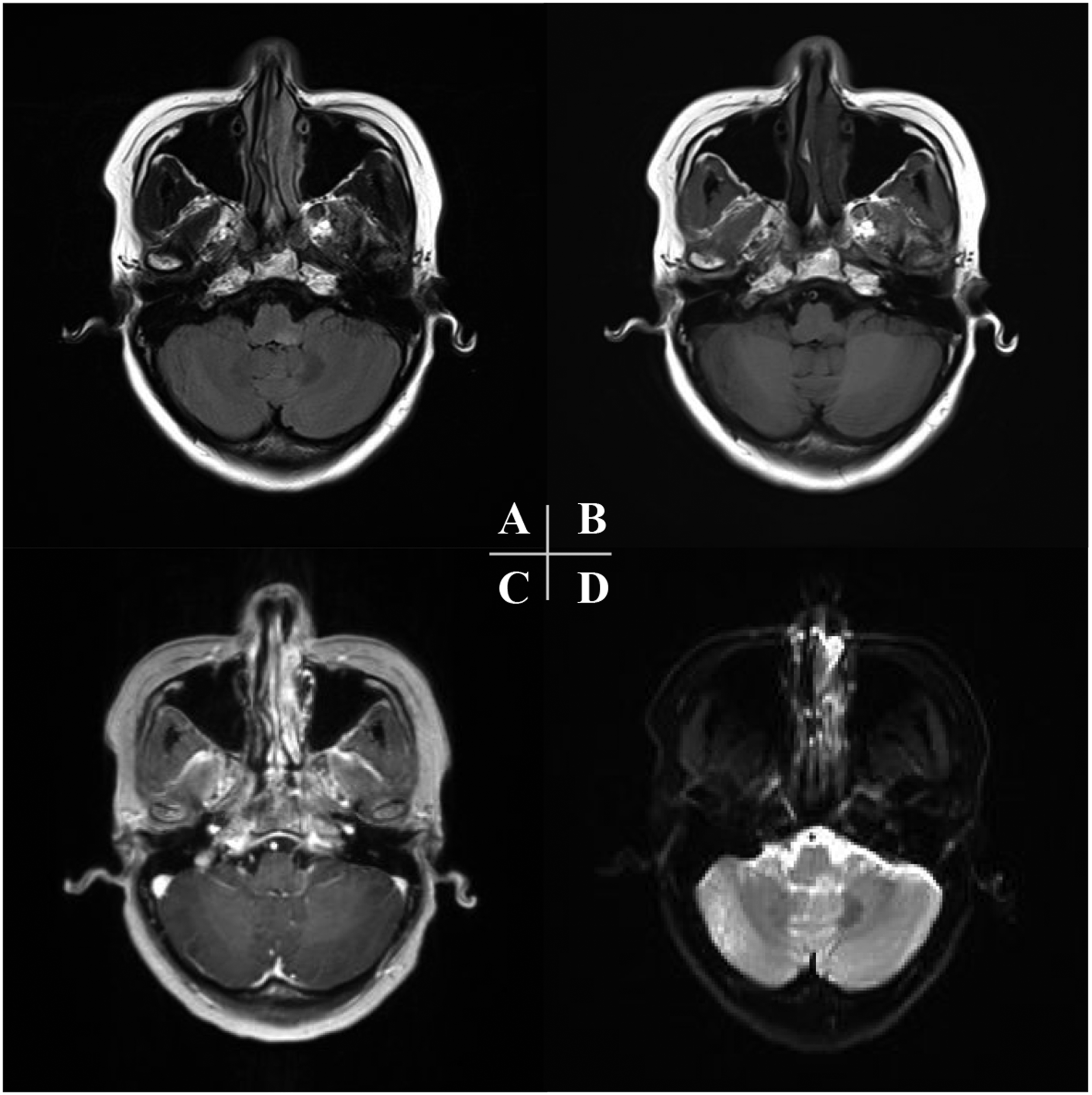
The patient suffered from rapid progression during the second week (Figure 2), including fever, blurred vision, dyspnea, left ptosis, diplopia, and drowsy consciousness. We gave her empiric ceftriaxone and acyclovir under the initial impression of viral encephalitis with brainstem involvement. Due to severe dysphagia, choking, and progressive tongue drop–related airway obstruction, she received emergent intubation and a tracheotomy for airway protection and better pulmonary toilet. Under the definite diagnosis as NMOSD on the 17th day, we started pulse steroids as methylprednisolone 1 g per day for 3 days and then tapered. Her vision loss, left-limb ataxia, vertigo, and limited tongue movement subsided, but her wide-based gait ataxia remained. Her spinal cord MRI showed no apparent abnormalities. Finally, she continued undergoing rehabilitation. Timeline of events in the 39-year-old patient diagnosed with neuromyelitis optica spectrum disorder who initially presented with vertigo. Abbreviations: ED, emergency department; 1st, first; 2nd, second; BID, twice daily.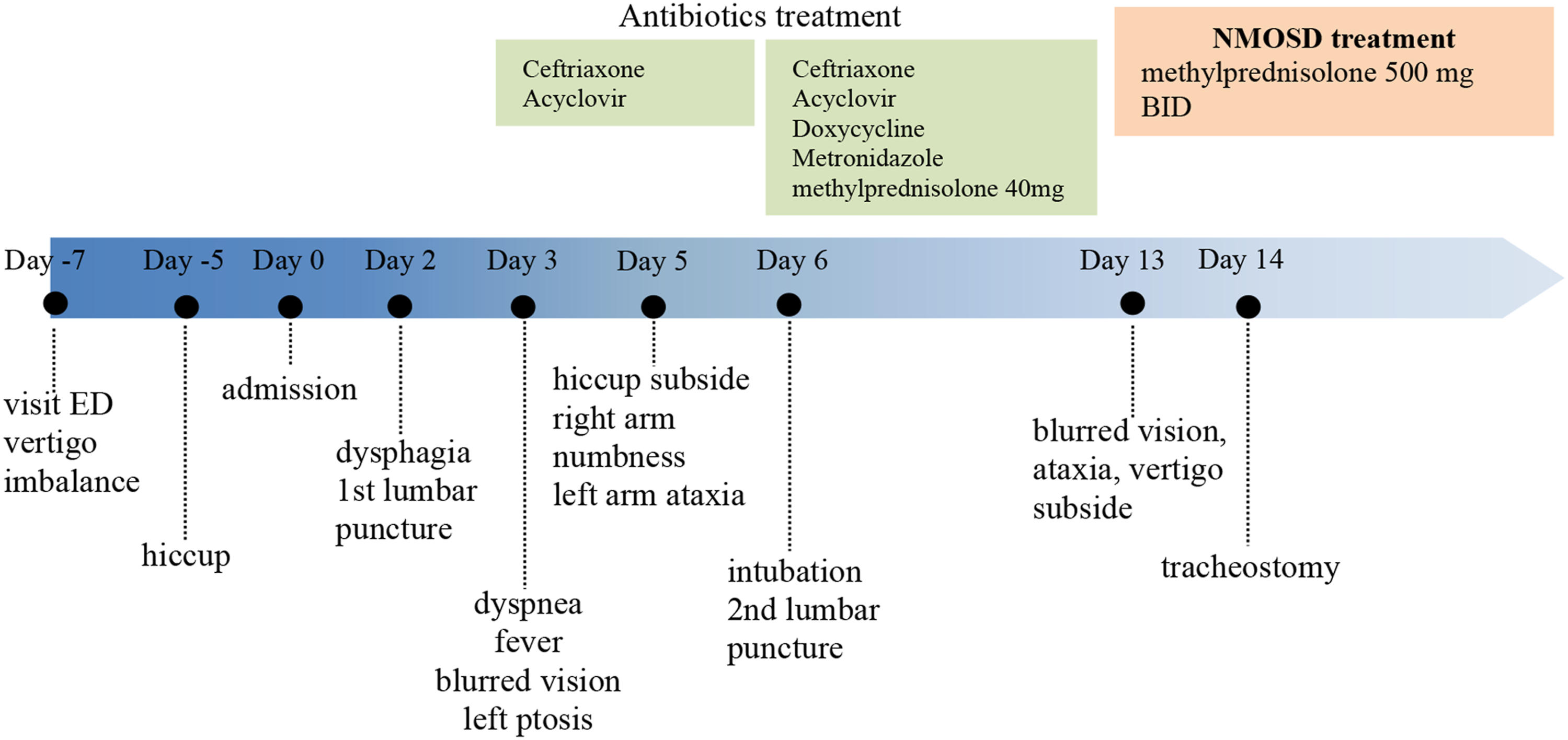
Discussion
NMOSD is a rare antibody-mediated inflammatory disease of the central nervous system. Its prevalence is less than 4.4/100,000 in the Western world, but has increased risk in individuals of Asian and African ancestry. 6,7 As in other autoimmune diseases, there is female predominance (ratio = 9:1), and the median age at presentation is 39 years. 8
We needed to confirm some critical points for prompt NMOSD diagnosis verification. First, intractable hiccups were the only sign that raised suspicion of a central nervous system lesion. According to the hiccup reflex arc, 9 the afferent impulse was carried by the vagus nerve, phrenic nerve, and sympathetic nerve and then processed by the upper spinal cord, medulla oblongata near the respiratory centers, the reticular formation, and the thalamus. Finally, the efferent impulse was carried by the phrenic nerve to the diaphragm. Any lesions that disturb the afferent, processing center, and efferent pathways can cause hiccups. We surveyed the central nervous system for intractable hiccups after excluding the cardiovascular, gastrointestinal, and pulmonary symptoms. Because the dorsal area of the medulla incorporates many components of this arc, a hemorrhagic or ischemic stroke, tumor, or inflammatory process involved in this area will be associated with persistent hiccups. Indeed, there was a faint patch of hyperintensity on our patient’s MRI T2-weighted images, which radiologists easily overlooked.
Second, marked serum leukocytosis with predominant segments and significant CSF lymphocytic pleocytosis put us in a dilemma. Although NMOSD usually follows an infection with related blood leukocytosis, 10 the patient denied any infectious signs except vertigo. In a study including 211 lumbar punctures from AQP4-Ab–positive NMOSD patients, 11 lymphocytic pleocytosis was detected in half of NMOSD patients, but it hardly exceeded 100 cells/uL. Therefore, we checked the AQP4-Ab first, but we still treated the patient initially as viral encephalitis, with empiric acyclovir and antibiotics even in the absence of detectable pathogens in the CSF.
Brainstem involvement is common in NMOSD, especially in the non-Caucasian population and AQP4-Ab seropositive cases. 12 Vomiting and hiccups are the most common brainstem manifestations (7%–46%) because the area postrema and the nucleus tract solitarius have a leaky blood–brain barrier, resulting in vulnerability to AQP4-Ab. Aside from the typical presentation, there are case reports about NMOSD mimicking meningoencephalitis with a fever of unknown origin. 13 Therefore, if we find a lack of improvement with antibiotics, combined with MRI findings and no detected pathogens, we must remember that the patient might have an NMOSD attack or exacerbation thereof.
Footnotes
Acknowledgments
The author would like to thank the radiologist Dr Wen-Ko, Su for brain and spine MR image interpretation. Besides, we would like to thank the anonymous (unknown) reviewers and the editor for their comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.