
Abstract
Cranial chordoma is a rare neoplasm that is challenging to both diagnose and manage. We present our experience in treating a case of clival chordoma mimicking a nasopharyngeal mass without any signs of cranial deficits or intracranial insult. Management was comprised of endoscopic transnasal debulking surgery followed by radiotherapy. However, we failed to achieve an oncologic outcome due to the development of lethal central nervous system and respiratory infections 1 month after the surgery.
Introduction
Chordoma are rare, locally invasive malignant tumors believed to originate from cell remnants of the primitive notochord. The annual incidence rate is approximately 0.8 per million people with male predominance. 1 Lesions commonly occur along the craniospinal axis, most frequently in patients in the fifth decade of life. The presentation of lesions varies with involvement site. In previous studies, cranial chordoma accounted for 32% of all chordoma cases. 1 The ventral extension of cranial chordoma may present as a nasopharyngeal or sinonasal mass. Intracranial progression of lesions can cause various neurologic symptoms. In computed tomography (CT) findings, intracranial progression of lesions is usually presented as a centrally located, expansile, destructive soft tissue lesion with possible intratumor calcification. 2 Obtaining a definite diagnosis is reliant on pathological study, and immunohistochemistry staining is essential to the process.
Because it frequently involves the skull base, treatment of cranial chordoma can be challenging. The relative anatomical inaccessibility makes surgical removal with an oncologically safe margin difficult. Thus, debulking surgery followed by adjuvant radiotherapy is suggested for better outcomes. 3 However, local recurrence remains a key problem due to the potential for incomplete tumor removal. Studies on achieving better management outcomes are ongoing. We present a case with timely diagnosis and good short-term surgical outcome.
Case report
A 50-year-old man presented with worsening nasal obstruction and blood-tinged nasal discharge. His medical record was unremarkable. No headache, visual impairment, or sign of cranial nerve palsy was present.
The man was referred to the medical center because of a large, grayish nasopharynx tumor completely obstructing the bilateral choanae (Figure 1A). Computed tomography scans with and without contrast (Figures 1B–1D) revealed a soft tissue lesion occupying the nasopharynx and posterior nasal cavities with intracranial extension to retroclival space. Bony destruction of clivus, sphenoid bone, sella floor and sphenoid sinus wall were present. Irregular intratumoral calcifications were also observed. The posterior margin of the tumor was somewhat close to the basilar artery. (A) Greyish nasopharyngeal lesion observed from left choana. (B) CT with contrast, axial view, illustrating posterior margin of the tumor close to the basilar artery (yellow arrow). (C) CT with contrast, coronal view. (D) CT with contrast, sagittal view, illustrating intracranial extension.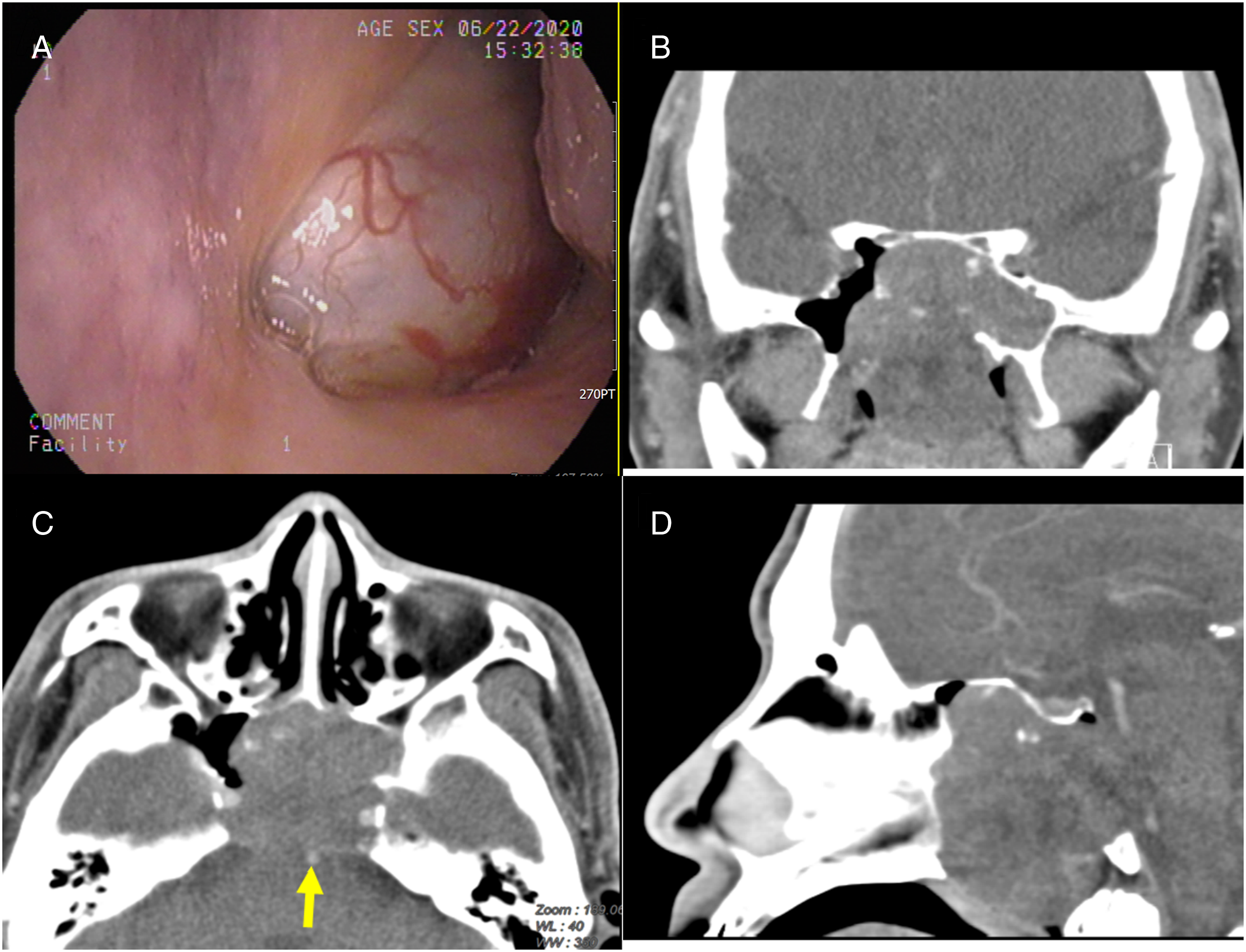
The patient underwent endonasal endoscopic tumor excision. Intraoperative frozen section pathology was performed, and pathologists identified the mass as chordoma. The differential diagnosis included chondrosarcoma or salivary gland malignancy. Literature
2,3
suggests surgical excision due to its appropriateness as a treatment for any of these tumors. Therefore, careful endoscopic debulking of the tumor was performed. However, the exposed dura began to leak cerebrospinal fluid (CSF). A free septal mucosal graft was harvested for repair of the CSF leak (Figures 2A, 2B). (A) Delicate debulking of tumor. (B) free mucosal graft (yellow arrow) from left nasal septum, used for repair of CSF leak. (C) H & E stain 100× magnification (physalipherous cells in stroma). (D) H & E stain 100× magnification (focal chondroid stroma). (E) H & E stain 400× magnification (physalipherous appearance). (F) EMA immunostaining.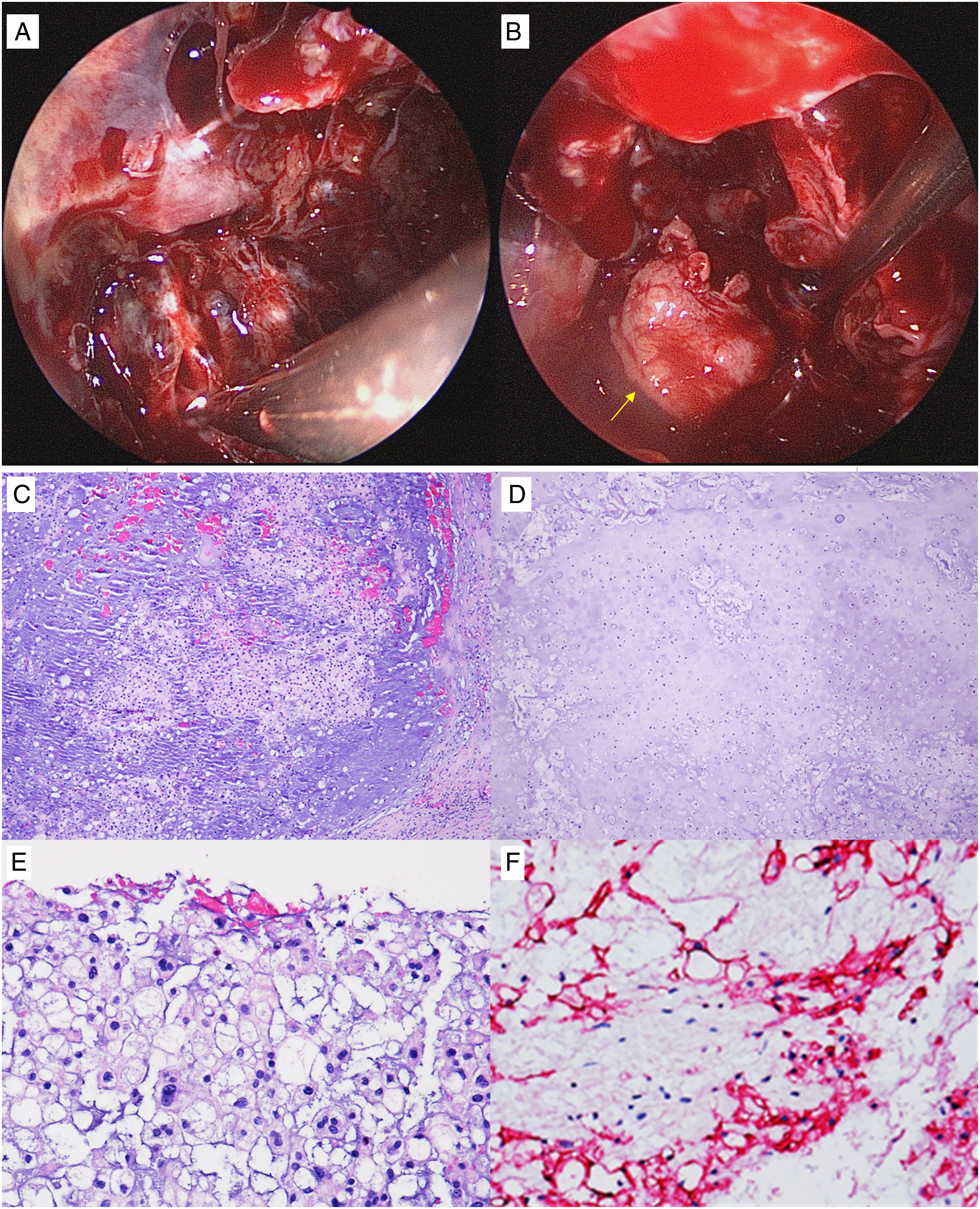
Histologically, the tumor was composed of lobules and cords of tumor cells with a physaliphorous appearance with abundant pale to eosinophilic cytoplasm with intracytoplasmic vacuoles. The tumor cells were in a myxoid and focal chondroid stroma, separated by fibrous septa. The tumor exhibited positive immunostaining for brachyury, cytokeratin (CK), and epithelial membrane antigen. The tumor was focally positive for S-100 and negative for p63 and D2-40. (Figure 2C-2F). Hence, a diagnosis of clival chordoma, chondroid subtype was established.
For the first month of postoperative recovery, the patient exhibited no signs of CSF leakage, CNS infection, or cranial nerve palsy. Radiation therapy was initiated 5 weeks after operation. However, after the third week of radiation therapy, fever with consciousness disturbance developed. The patient expired due to lethal central nervous system and respiratory infections.
Discussion
Cranial chordoma is rare, and presentation can be variable based on anatomical involvement. This renders diagnosis of cranial chordoma challenging. Our case reported only nasal blockage caused by the ventral extension of the tumor. Despite having exhibited dorsal and lateral tumor extension, the cranial chordoma did not cause headache or any cranial neuropathy. We emphasize that cranial chordoma’s neurologically silent presentation can potentially hinder a timely diagnosis, unless early fiberoptic examination or further image study is performed.
Chordoma is classified into three histological subtypes: conventional, chondroid and dedifferentiated. 4 Pathognomonic physaliphorous (vacuolated appearance) cells within myxoid stroma are typically observed in the conventional subtype. 5 Our case belongs to the chondroid subtype, which is characterized by the presence of a chondroid differentiation area within the conventional chordoma tissue. 6 The findings of previous studies suggest no notable difference in survival rate between the conventional and chondroid subtypes. Although female predominance and younger age have typically been observed in chondroid subtype, 7 neither of these were present in our case. High-grade sarcoma exists in the dedifferentiated subtype, which is associated with the worst outcomes.
The principle to treat chordoma is to optimize surgical tumor resection with subsequent adjuvant (mostly radiation) therapy. A cranial chordoma can be close to, or can even invade, vital structures, such as the cranial nerves, pituitary gland or brain. Experienced surgeons must both effectively eradicate the tumor and preserve the nearby structures of the skull base. Many surgical approaches have been utilized, including endoscopic transnasal, transotic and lateral skull base approaches. 8 Because the tumor of our case was centrally located and was accessible from bilateral choanae, we applied an endonasal endoscopic approach. The rate of intraoperative CSF leakage in endonasal endoscopic surgery for chordoma is approximately 50%. Multiple-layer repair is usually required because the CSF leakage events in chordoma surgery are commonly high-flow. 9
This experience in treatment of clival chordoma revealed that timely diagnosis can be somewhat difficult due to its insidious onset and neurologically silent presentation. Our patient presented with only nasal obstruction initially. However, the lesion had already invaded the retroclival region. This difficulty may lead to higher surgical risks and a higher local recurrence rate due to potentially incomplete excisions. Thus, clinicians must note that clival chordoma, although rare, should be kept in mind in differential diagnosis of nasopharyngeal mass.
Conclusion
Clival chordoma, rare and insidious, can mimic a nasopharyngeal mass. Skull base surgery can be challenging, and caution is essential in preoperative evaluation and mitigation planning.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.