
Abstract
We report a 6-year-old girl with progressive bilateral conductive hearing loss for 2 years. She passed the newborn hearing screening conducted with otoacoustic emissions testing and had a normal development of speech and language, which indicated that her deafness was delayed-onset. She also had congenital proximal interphalangeal joints. Proximal symphalangism was confirmed by genetic testing (NOG gene: c.406C > T, p.R136C). Bilateral stapes ankyloses were proved by surgery and her hearing was improved after stapedotomy by over 30 dB. Besides, this case should remind clinicians to carefully distinguish NOG gene-related deafness from congenital ossicular malformation and pediatric otosclerosis.
Introduction
The NOG gene plays an important role in the course of bone and joint development. Mutations in the NOG gene with autosomal dominant inheritance can cause a variety of syndromes involving malformations of the bones and joints, which are known as NOG-related symphalangism Proximal symphalangism (SYM1) spectrum disorders (NOG-SSDs). Symmetrical moderate bilateral conductive deafness is a common manifestation in some NOG-SSDs, including SYM1, multiple synostoses syndrome 1, and stapes ankylosis with broad thumbs and toes (SABTT). 1 NOG-related hearing loss, mainly caused by the fixation of the stapes footplate, may be the major symptom in some sporadic mild cases with unremarkable general abnormalities; this symptom is easily confused with symptoms of congenital ossicular malformation (COM) or otosclerosis, especially in children. In addition, the timing of the onset of hearing loss remained unclear in most of the previously reported pediatric cases of NOG-SSDs. Here, we report a sporadic case of SYM1 in a patient who had normal hearing at birth, was noted to have progressive hearing loss from 4 years old, and underwent bilateral surgeries with satisfactory hearing improvement at 6 years old. The case reminds otologists of the possibility of NOG-SSDs when encountering children with conductive deafness and provides theoretical support for the notion that NOG gene-related syndromic conductive deafness can occur and become aggravated during childhood despite normal hearing at birth. This research was exempt from the Institutional Review Board of Peking Union Medical College Hospital (S-K1121). The parents of the patient provided written consent.
Case Report
A 6-year-old girl was referred to the Peking Union Medical College Hospital for treatment of bilateral hearing loss. The parents noticed that the child’s hearing had been mildly decreasing for 2 years. The child passed the newborn hearing screening with otoacoustic emissions, and no significant difference was observed in her speech and language development compared with her peers. No history of otitis media or trauma was noted. No other family members with similar symptoms were identified.
Physical examination showed jug-shaped pinnae with normal ear drums. Bilaterally, her fifth fingers were significantly shortened and inflexible (Figure 1A and B), a finding that had been identified at birth. Hypermetropia had recently been diagnosed and was corrected with glasses, and no other significant physical malformations were observed.
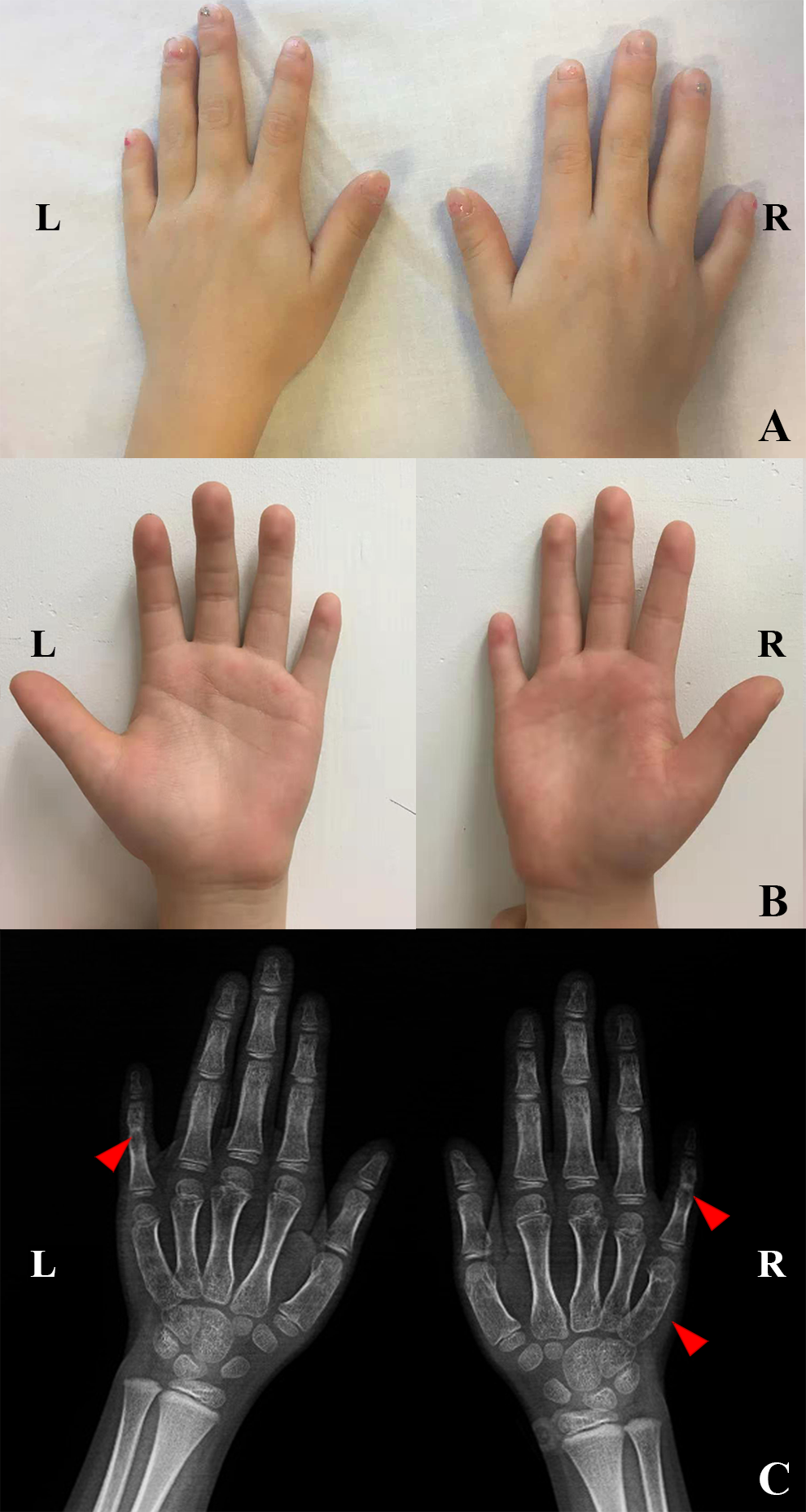
A and B, The bilateral little fingers are significantly shortened and unable to flex. The shape and activity of the remaining fingers are normal. C, Red arrows indicate fusion of the proximal interphalangeal joints of the fifth fingers of both hands on the films, and the fifth metacarpal of the right hand is short.
Pure tone audiometry revealed bilateral moderate conductive deafness (Figure 2A). Gelle’s tests of both ears were negative. High-resolution computed tomography of the temporal bone showed no significant abnormalities (Figure 2B). X-rays showed fusion of the proximal and middle phalanges of the bilateral fifth fingers and a shortened fifth metacarpal of the right hand (Figure 1C).
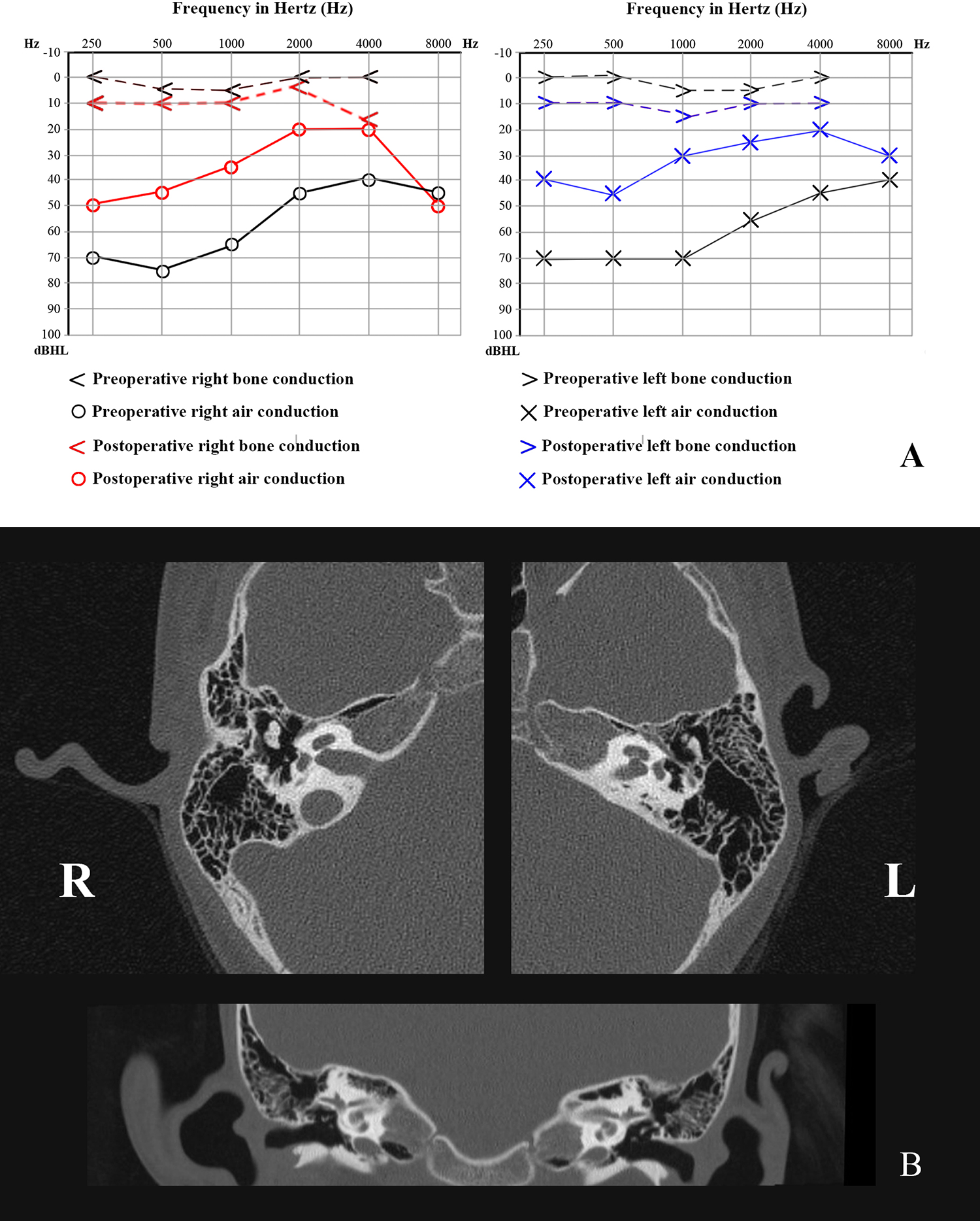
A, Pure tone audiometry revealed moderate conductive deafness in both ears and the average air-bone gap of both ears was improved by 38.75 dB in the left ear and 33.5 dB in the right ear. B, High-resolution computed tomography of the temporal bones (axial and coronary slices) did not show any anomalies.
A NOG gene mutation (c.406C > T, p.R136C) was confirmed by genetic testing. The mutation was heterozygous, and both of her parents were negative for the mutation.
The patient underwent successive surgeries for both ears with an interval of 4 months. During the surgeries for both ears, the malleus and incus were confirmed to have normal shape and mobility, but the anterior and posterior crura of the stapes were thickened, the annular ligaments were sclerotic, and the footplates were fixed. Therefore, fenestration of the stapes footplate and implantation of a piston-like ossicular prosthesis were performed to restore hearing. The patient’s hearing thresholds were >30 dB improved in both ears and remained stable during the 1 year of follow-up.
Discussion
Conductive hearing loss in NOG-SSDs can be diagnosed during childhood, but few reports have provided information on hearing capacity at birth, and the onset of deafness currently cannot be accurately determined. In this case, the child passed the newborn hearing screening tests and had good speech and language development after birth. The hearing loss was not noticed until she was 4 years old, indicating normal hearing and middle ear function at birth and confirming that her progressive hearing loss developed during childhood. This report serves as a good data supplement for the notion that conductive deafness caused by SYM1 not only is congenital 1 but can also be delayed-onset, 2 which has rarely been reported.
NOG gene-related conductive deafness can be easily misdiagnosed as COM or otosclerosis due to the similar manifestations of these conditions. The main differences among these diseases are as follows: (1) The onset of conductive deafness associated with NOG-SSDs and COM occurs much earlier than that of otosclerosis 3,4 or juvenile otosclerosis. 3 (2) Congenital ossicular malformation and otosclerosis can present with ipsilateral deafness, while NOG-SSDs are always associated with bilateral deafness. 4,5 Unlike COM and otosclerosis, NOG-SSDs are often accompanied by other bone and joint abnormalities, as in the case reported here. 6 (3) Otosclerosis lesions are located at the stapes footplate and extend to the bony wall of the labyrinth. 7 However, NOG-SSDs and COM mainly cause bony abnormities of the ossicular chain, especially the stapes footplate, 1,8 such as the hyperosteogenic stapes in the present case. High-resolution computed tomography of the temporal bone can help to detect anomalies of structures in the ear. (4) NOG-SSDs are commonly observed in families carrying mutations in the NOG gene, but sporadic cases, such as in the present case, can also occur. 9 Otosclerosis is an autosomal dominant disease with 20% to 40% penetrance. 10 However, COM usually occurs sporadically. 11 Accordingly, for children with bilateral conductive deafness, stapes ankylosis, otosclerosis, and COM should be differentiated from NOG-SSDs.
The human NOG gene is located on chromosome 17q22, and its translated noggin protein is a secreted antagonist of bone morphogenetic proteins (BMPs), which are crucial for normal bone and joint formation. 12 The currently known structural changes in noggin caused by NOG gene mutations mainly occur in 3 regions: (1) the binding region of type I or type II receptors of BMPs, where changes affect the binding of noggin to BMPs; 13,14 (2) the terminal hydroxyl groups on noggin, where changes affect the formation of active homodimers of noggin; 15 and (3) the heparan region, which helps the noggin-BMP complexes cling to the cell membrane. 16 In this case, genetic testing confirmed that the patient had a heterozygous missense mutation (c.406C > T p.R136C) in the NOG gene, resulting in substitution of the positively charged arginine with a neutral cysteine in the heparan region of the noggin protein. The binding affinity of noggin-BMP complexes to the cell membrane was decreased, causing an increase in the concentration of local BMPs and thus leading to dysplasia. A case with the same locus has been reported previously. 17
Conclusion
We reported a sporadic case of SYM1 with delayed-onset bilateral conductive hearing loss that was treated with bilateral stapedotomy, which yielded satisfying hearing results. This case suggests that NOG gene-related deafness can occur in childhood with normal hearing at birth. Future long-term follow-up with serial hearing tests for this girl will be needed to fully understand the development process of SYM1-related hearing loss, as patients with other skeletal disorders may develop a sensorineural hearing loss secondary to otic capsule demineralization. Family history, general physical examination, and genetic analysis can help to distinguish NOG gene-related syndromes from otosclerosis and COM.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81300830).