
Abstract
Background:
Otosclerosis is a focal lesion of the inner ear. The role of genetic factors in the pathogenesis of otosclerosis has received increasing attention. We analyzed the clinical manifestations, inheritance pattern, and pathogenic genes in a family with otosclerosis.
Methods:
We collected clinical data and generated a family pedigree. High-throughput second-generation sequencing technology was used to identify candidate genes by performing whole-exome sequencing of 7 members of the family, and Sanger sequencing was performed to validate candidate gene mutations in the 7 family members.
Results:
Otosclerosis was characterized by autosomal dominant inheritance in this family. Whole-exome sequencing did not reveal mutation sites in known deafness-related genes. However, a c.2209A > G (p.T737A) mutation was detected in exon 6 of the SP1 gene, which is associated with the COL1A1 gene. This mutation was a pathogenic mutation, and Sanger sequencing confirmed that this mutation cosegregated with the clinical phenotype among the family members.
Conclusions:
The pattern of otosclerosis in this family is consistent with autosomal dominant inheritance, and the SP1 gene, harboring the c.2209A > G (p.T737A) mutation in exon 6, may be the causative gene of otosclerosis in this family.
Introduction
Otosclerosis is a focal lesion of the bony labyrinth with an unknown cause. In the bony labyrinth, new spongy bone locally replaces the normal bone, after which the new bone undergoes ossification and hardening. The pathological changes are mainly related to abnormal bone remodeling. If the lesion invades the stapedial annular ligament, causing fixation of the footplate of the stapes or restriction of its movement, then conductive hearing loss will occur. If the lesion invades the cochlear region or the internal auditory canal, then sensorineural hearing loss will occur. The main clinical manifestation in patients with otosclerosis is progressive hearing loss, which may be accompanied by tinnitus and dizziness. 1,2 The incidence of otosclerosis is higher in Caucasians, at 0.3% to 0.5%, than in Asian and black populations. Otosclerosis does not have a recognized pathogenesis, and endocrine, immunological, viral infection, and genetic theories have been postulated to explain the etiology of otosclerosis. 3 In recent years, the role of genetic factors in the pathogenesis of otosclerosis has received increasing attention. 4 -6 According to many scholars, 7 -10 otosclerosis is characterized by autosomal dominant inheritance accompanied by incomplete dominant inheritance, but some scholars have also suggested the possibility of chromosomal recessive inheritance, and an effect of the interaction between X-linked dominant inheritance and autosomal recessive inheritance on its penetrance cannot be excluded. Currently, genes known to cause otosclerosis include OTSC1, OTSC2, OTSC3, OTSC4, OTSC5, OTSC7, OTSC8, OTSC10, COL1A1, TGFB1, PPP2R5B, BMP2, and RELN. 5 -11
In this study, otological, audiological, and genetic characteristics were analyzed in a Chinese family with clinically diagnosed otosclerosis. Whole-exome sequencing and Sanger sequencing were used to identify pathogenic genes in this family, and the preliminary findings revealed that the SP1 gene, carrying the c.2209A > G (p.T737A) mutation in exon 6, may be the causative gene of otosclerosis in this family, as described below.
Patients and Methods
Ethics Approval
The survey and sampling of family information were approved by the Ethics Committee of Peking Union Medical College Hospital and Ethics Committee of Basic Medical Sciences, Chinese Academy of Medical Sciences (No. 003-2014). Informed consent was obtained from the family members.
Family Data
The participants of this study were members of a Chinese family with otosclerosis who were treated at the Peking Union Medical College Hospital, and their medical histories and otological and audiological information were collected to generate a family pedigree (Figure 1). All included members completed a patient information registration form, with which the following detailed information was collected: personal information, presence or absence of hearing loss, age of onset of hearing loss, predisposing factors, clinical symptoms, progression of hearing loss, history of consanguineous marriage, history of ototoxic drug use, history of head trauma, pregnancy, and history of jaundice in infancy. At the same time, all the included family members (except II5, who was not examined for physical reasons) were subjected to a detailed systemic examination, an otological examination, an audiological examination, and blood sampling for genetic testing.
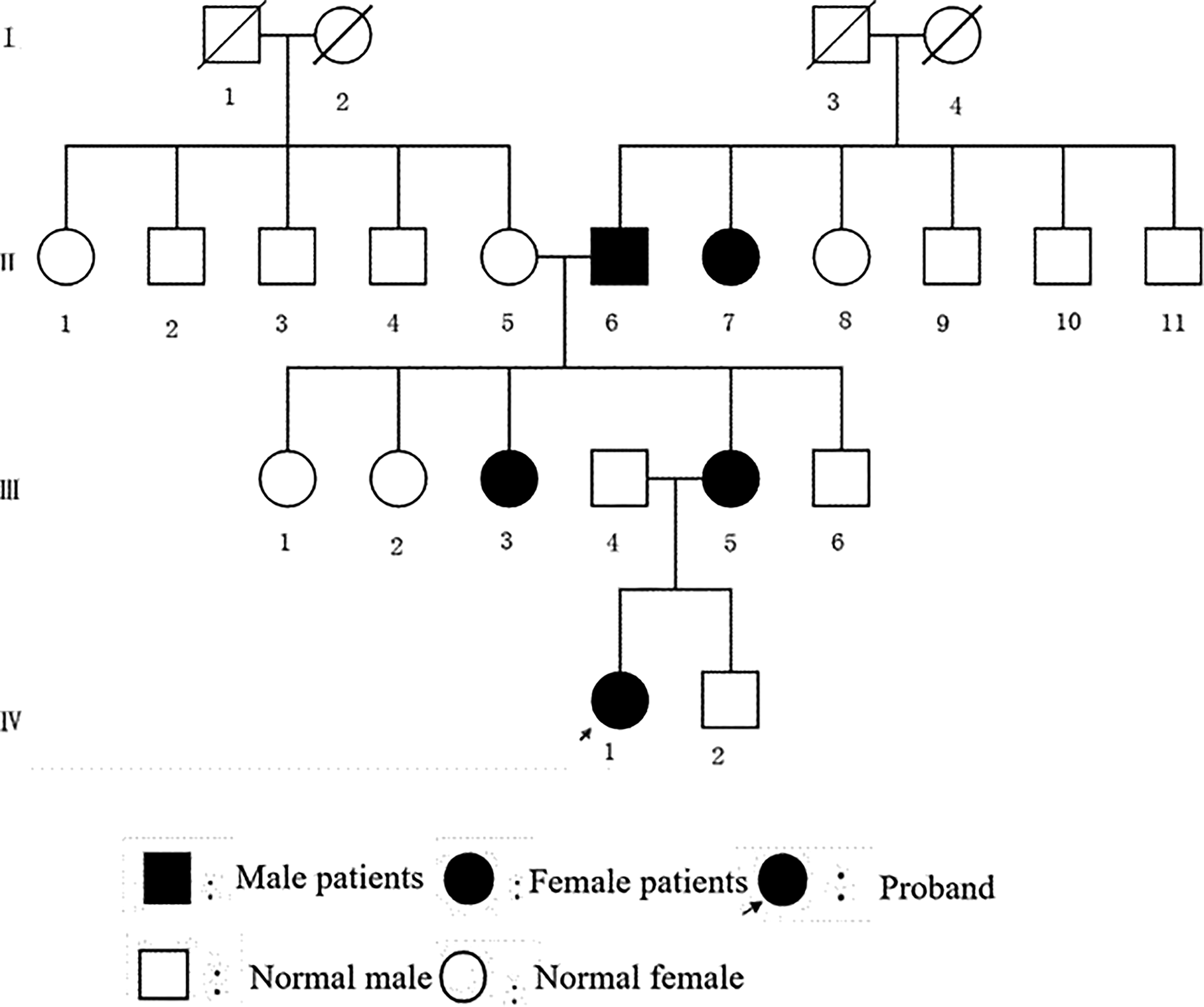
The family pedigree chart. This family has 23 individuals in 4 generations. II6, II7, III3, III5, and IV1 were diagnosis of otosclerosis, IV1 was the proband who was an 18-year-old female, and II5 and III4 were without otosclerosis.
Clinical Audiological Examination and Imaging Examination
Pure-tone audiometry and the Gellé test were performed on the included family members using a Conera pure-tone audiometer (Madsen; GN, Ballerup, Denmark) to preliminarily determine the degree of hearing loss and ossicular chain activity. An acoustic immittance test was performed using an OTOflex 100 acoustic immittance meter (GN) to determine the condition of the middle ear. A distortion product otoacoustic emission examination was performed using an Eclipse ear dynamic analyzer system (Interacoustics, Denmark) to determine the function of the outer hair cells of the inner ear. A computed tomography (CT) scan of the temporal bone of the proband (IV1) and 2 other family members with deafness (II7 and III3) was performed using a Siemens 64-slice spiral CT instrument (Somatom Sensation 64; Siemens AG Medical Solutions, Forchheim, Germany) to determine the presence or absence of organic lesions in the ear.
Analysis of the Pattern of Inheritance
Based on the presence or absence of concomitant diseases, hereditary deafness is divided into syndromic and nonsyndromic deafness. The inheritance of nonsyndromic deafness is divided into autosomal dominant inheritance, autosomal recessive inheritance, sex chromosome-linked inheritance (including X-linked dominant inheritance, X-linked recessive inheritance, and Y-linked inheritance), and mitochondrial maternal inheritance. We analyzed the inheritance pattern of otosclerosis in this family.
DNA Extraction and Whole-Exome Sequencing
Five milliliters of peripheral blood was collected from the 7 family members (II5, II6, II7, III3, III4, III5, and IV1) who participated in this study. DNA was extracted according to the method described in the instruction manual of the Genomic DNA Extraction Kit (CWBio), and a DNA library was constructed. GenCap liquid-phase target gene capture technology (MyGenostics Inc, Beijing, China) was used to detect whole exomes, including more than 200 known deafness genes, such as the otosclerosis-associated genes OTSC1-10, COL1A1, TGFB1, PPP2R5B, BMP2, and RELN, and other common causative genes of deafness, including GJB2, GJB3, SLC26A4, and OTOF. An Illumina NextSeq 500 high-throughput sequencing system was used to obtain raw data.
Analysis of Sequencing Results
Raw sequences from sequencing reactions were aligned to the human genome using the Burrows-Wheeler Aligner after removing adaptors and low-quality sequences. Variant detection of single-nucleotide polymorphisms (SNPs) and insertion and deletion (indel) mutations was performed with GATK software, and multiple databases (such as dsSNP, 1000G, ESP6500, HGMD, and OMIM) were simultaneously searched using ANNOVAR software(version, 2014-11-12). From the pathogenic analysis, the pathogenic mutation sites (pathogenic), mutant bases that had been sequenced more than 5 times, sites with a mutation frequency greater than or equal to 30%, and sites that were not present or had a mutation frequency less than 5% in the 5 mutation databases of healthy people, including 1000 g2015apr, ESP6500si, Inhouse, ExAC-ALL, and ExACEAS, were identified. In addition, the mutation sites and pathogenic mutation sites that had been reported in the literature were retained, while the synonymous mutation sites in the data set were removed. A statistical analysis of SNPs and indels was performed. MutationTaster, PolyPhen, and SIFT software were used to predict the pathogenicity of the mutations, and after considering relevant publications, the most likely pathogenic genes and mutations were identified for further validation.
Sanger Sequencing Verification
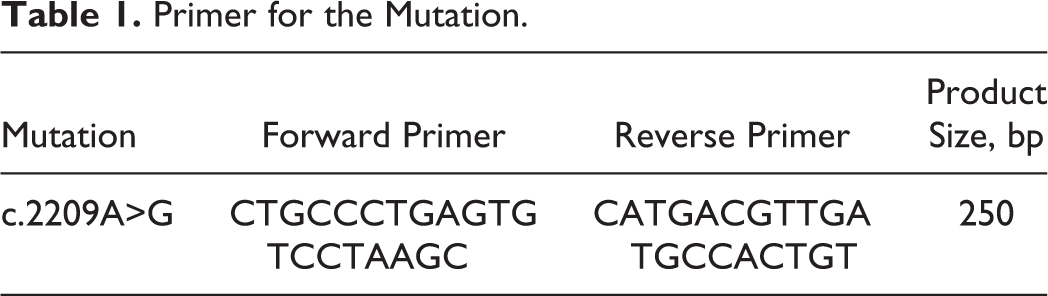
Primers were designed using Premier 5.0 software after querying the standard sequences of suspected pathogenic genes in the GenBank database (Table 1). Afterward, polymerase chain reaction amplification and Sanger sequencing (MyGenostics Inc) were used to analyze the cosegregation of suspected mutated gene loci in 5 members of the family.
Primer for the Mutation.
Results
Clinical Data From the Family With Otosclerosis
This family with otosclerosis includes 23 individuals in 4 generations (Figure 1), and members of the family have been living in Hebei Province for many years. In this study, 7 individuals (II5, II6, II7, III3, III4, III5, and IV1; Table 2) in the family were interviewed, 5 (II6, II7, III3, III5, and IV1) of whom were clinically diagnosed with otosclerosis and 2 (II5 and III4) of whom did not have otosclerosis. The proband (IV1) was an 18-year-old female. None of the participants in this study had a history of consanguineous marriage, ototoxic drug use, head trauma, jaundice in infancy, or otitis media. II6 and III5 didn’t perceive their hearing loss before taking a hearing test; other 3 of the patients (II7, III3, and IV1) with otosclerosis presented progressive hearing loss and occasional tinnitus in both ears, without discomfort such as vertigo, otorrhea, headache, or vomiting. Neither of the family members without otosclerosis (II5 and III4) had a history of hearing loss, tinnitus, vertigo, or otorrhea.
The Demographic Informations and Findings of the Individuals in the Family.

Specialized and Systemic Examinations
All included subjects underwent a specialized physical examination. No abnormalities were observed in auricle, the external auditory canal, or the tympanic membrane, and a systemic examination did not detect any other systemic lesions. The results of the pure-tone audiometry assessment revealed conductive hearing loss in both ears of all 5 patients with otosclerosis in this family, and the degree of hearing loss ranged from mild to severe, with an air-bone gap of 10 to 40 dB (Figure 2). The results of the Gellé test were negative in both ears. The high-resolution CT examination of the temporal bone (II7, III3, and IV1) showed thickening of the footplate of both stapes and patchy shadows with decreased density around the vestibular window (Figure 3). No abnormalities were detected (data not shown) in the specialized examination or hearing examination of the family members without otosclerosis. An initial diagnosis of otosclerosis was made based on the following observations: asymmetrical progressive hearing loss in both ears with an unknown cause, a normal tympanic membrane or positive Schwartz sign, a negative Gellé test, a type-A tympanogram, good Eustachian tube function, and a loss of the stapedius reflex. According to the abovementioned criteria, participants II6, II7, III3, III5, and IV1 in this family had otosclerosis, while participants II5 and III4 did not have otosclerosis.
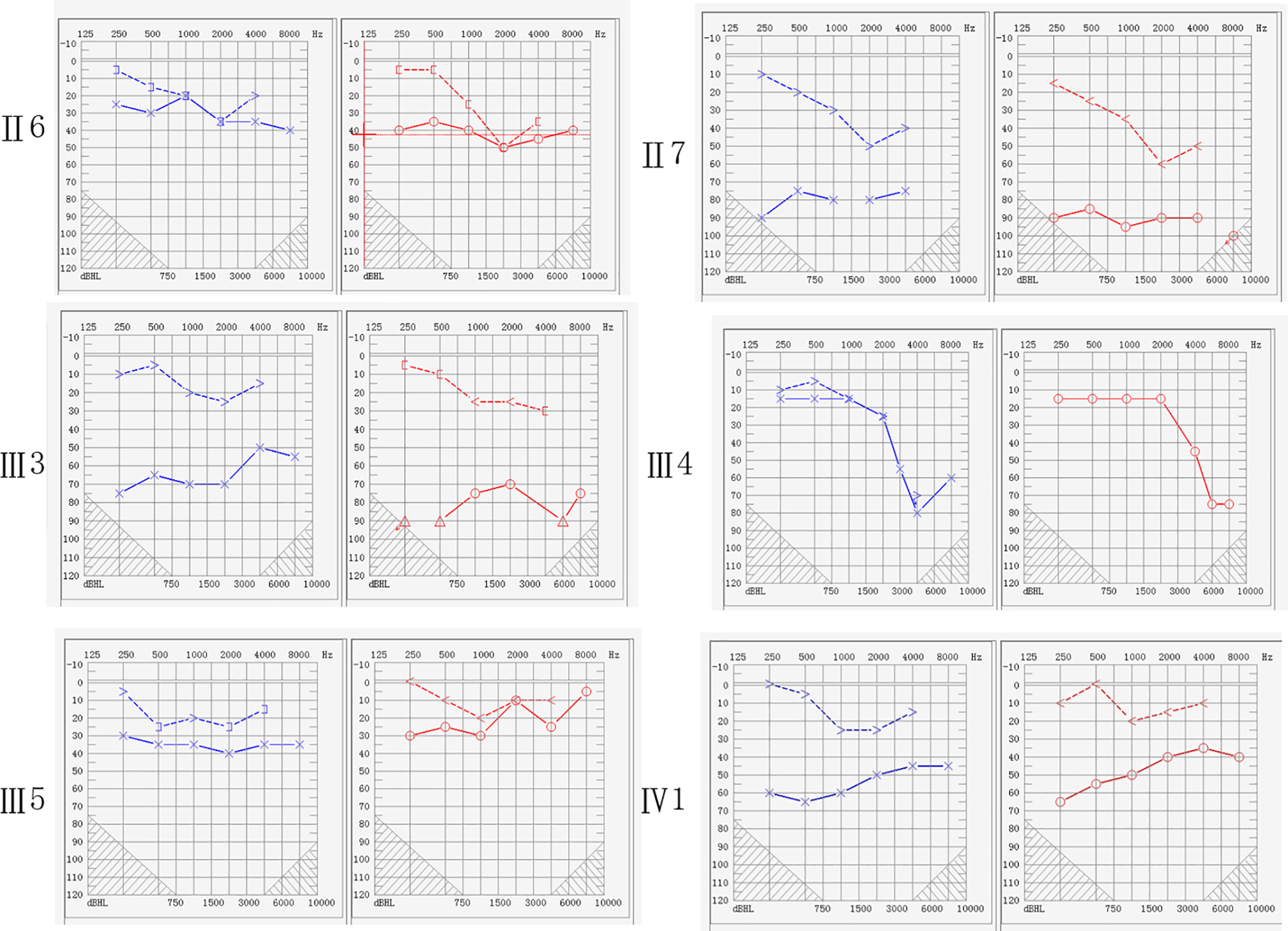
The pure-tone audiometry assessments. The results of the 5 patients with otosclerosis indicate conductive hearing loss, and the degree of hearing loss ranged from mild to severe, with an air-bone gap of 10 to 40 dB.
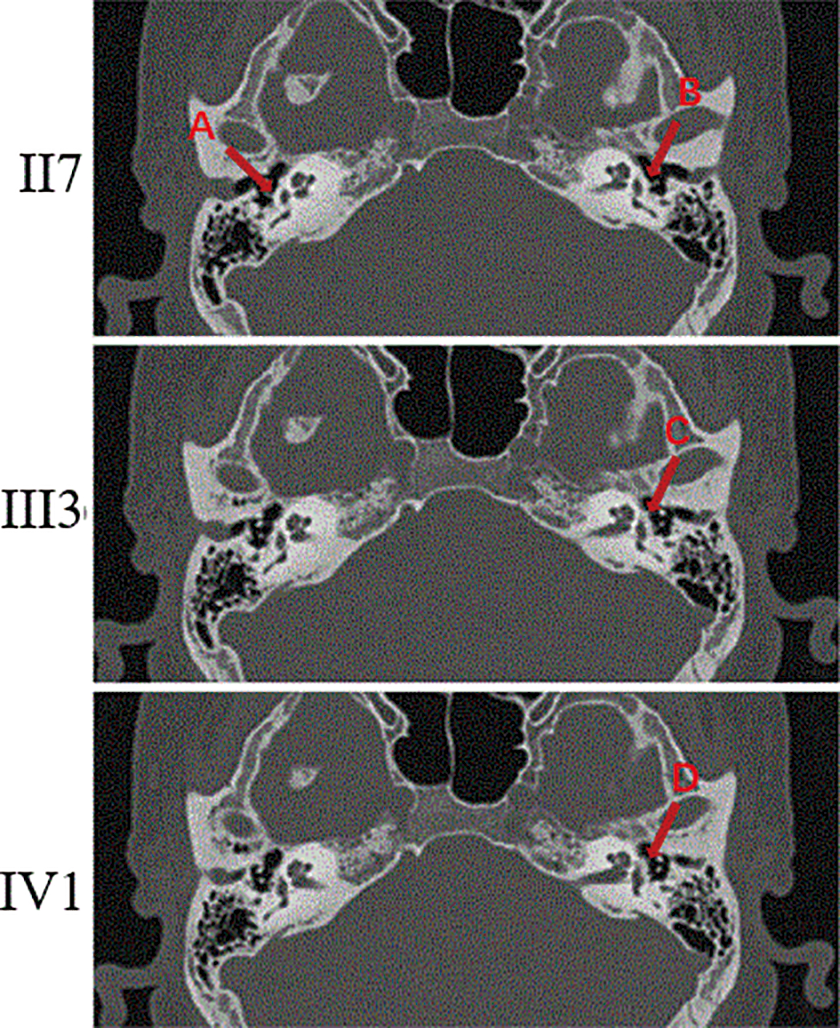
The high-resolution computed tomography (CT) examination of the temporal bone (II7, III3, and IV1). Red arrowheads of A and B showed thickening of the footplate and red arrowheads of C and D showed decreased density around the vestibular window.
Analysis of Genetic Characteristics
This family includes 4 generations, and each generation includes deaf patients with a clinical manifestation of conductive hearing loss in both ears. One of the parents of each patient was also a patient, and both the male and the female members of each generation developed the disease. The youngest and oldest ages of onset of the disease were 18 years and 65 years, respectively, and this family displayed characteristics consistent with autosomal dominant inheritance. The patients’ symptoms and the physical examination did not show any other accompanying systemic diseases; therefore, this family is considered to have nonsyndromic deafness.
Results of the Analysis of Whole-Exome Sequencing Data
In this family with otosclerosis, whole-exome sequencing, screening, and analysis did not detect any mutation sites in common deafness-related genes or otosclerosis-related genes. However, a heterozygous base mutation site, c.2209A > G (p.T737A; Table 2), was detected in the SP1 gene, which is associated with the otosclerosis-causative gene COL1A1. This mutation site was located in exon 6 of the SP1 gene and was a nonsynonymous mutation. The allele frequency of this mutation was less than 1% in the ESP, 1000 Genomes Project, and EXAC databases. The results obtained from MutationTaster and MutationTaster_Predict were “0.902” and “Disease_causing,” respectively, but the results obtained from the PNo conscious hypoacusisolyPhen2 and SIFT analyses were “Benign” and “0.217,” respectively. Sanger sequencing detected this mutation site in all patients in this family, but the site was not detected in the family members without otosclerosis (Figure 4). Based on the Sanger sequencing results, this mutation site cosegregated with the clinical phenotype.
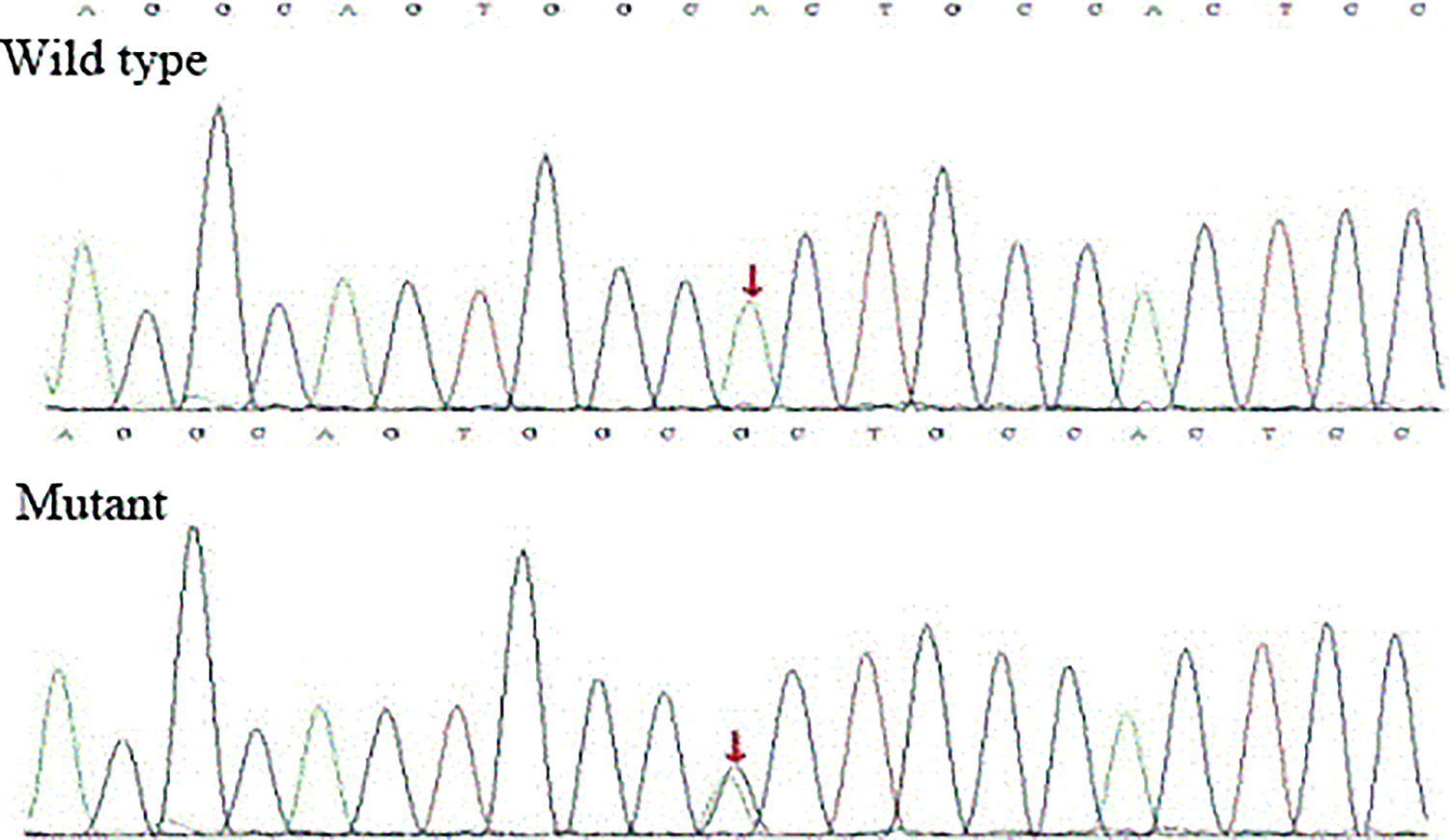
The Sanger sequencing results of the family. The mutation site can be detected in all patients in this family, but it was not detected in any family members without otosclerosis.
Discussion
Otosclerosis is a focal lesion that originates in the otic capsule. Many hypotheses 12,13 have been proposed to explain the etiology of otosclerosis, in which genetic factors play an important role in pathogenesis. In this study, data were collected from a family with otosclerosis. The family pedigree obtained with genetic analysis was consistent with the characteristics of autosomal dominant inheritance, and a c.2209A > G (p.T737A) mutation in the sixth exon of the SP1 gene was identified that may be the causative mutation of otosclerosis in this family. This mutation cosegregated with the otosclerosis phenotype among the family members and may be a new pathogenic mutation for otosclerosis.
This study revealed that genetic factors are related to the pathogenesis of otosclerosis, which is similar to the findings of other studies. In 1861, Toynbee postulated that otosclerosis may be related to genetic factors. Later, Albrecht, Larsson, and other researchers 14 -16 proposed that otosclerosis was characterized by autosomal dominant inheritance and possibly incomplete dominance. In 1998, Tomek discovered and reported the first otosclerosis-associated gene, OTSC1. Following the discovery of this gene, more than 10 otosclerosis-associated genes, including OTSC2-OTSC10, COL1A1, TGFB1, PPP2R5B, BMP2, and RELN, have been successively discovered and reported. 17 -19
In the present study, we identified a mutation site in the SP1 gene, which is associated with COL1A1 gene transcription. McKenna et al 20 identified an association between a polymorphism in the SP1-binding site in the COL1A1 gene and otosclerosis. The SP1 gene encodes SP1 protein, which belongs to the SP protein family and is a common transcription factor. An SP1 transcription factor-binding site is present in the first intron of the COL1A1 gene. Mutation of the first intron of the COL1A1 gene affects the affinity of the SP1 protein for the COL1A1 gene and then affects the transcription of the COL1A1 gene. The COL1A1 gene encodes the α1 chain of type I collagen, which is one of the important proteins comprising bone. Abnormal expression of the COL1A1 gene affects the formation of type I collagen and subsequently causes bone abnormalities. We propose that the possible pathogenic mechanism of the mutation site in the SP1 gene, leading to otosclerosis is an effect on the affinity of the SP1 transcription factor for the SP1-binding site in the first intron of the COL1A1 gene, which in turn affects the transcription of the COL1A1 gene and the formation of type I collagen and ultimately causes abnormalities in the stapes and pericochlear bone, leading to the development of otosclerosis.
The study identified a c.2209A > G (p.T737A) mutation in the sixth exon of the SP1 gene related to otosclerosis for the first time, which provides evidence to support genetic counseling and prenatal diagnosis for this family and will help further reduce birth defects. Our study also enriches the gene spectrum of otosclerosis and provides new candidate sites for gene-based diagnosis of otosclerosis.
Our study also has limitations. At present, there is no direct evidence for the molecular mechanism of otosclerosis caused by SP1 gene mutation. In the future, we will continue to follow-up with this family to provide a theoretical basis for further exploration of the genetic mechanism of otosclerosis and the study of the clinical characteristics of patients with deafness in families with otosclerosis. Another limitation is the small sample size. Examinations of families with larger numbers of members with otosclerosis is needed.
In conclusion, otosclerosis is characterized by autosomal dominant inheritance in this family. No mutation sites were identified in known deafness-related genes, and the c.2209A > G (p.T737A) mutation in the sixth exon of the SP1 gene may be the causative mutation of otosclerosis in this family.
Footnotes
Authors’ Note
Y. Zhang and Q. Tang contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grands of the Beijing Natural Science Foundation (No. 7172176), National Natural Science Foundation of China (NO.81470698), and National key research and development plan of China (NO.2016YFA0101000, NO.2016YFA0101002).