
Abstract
Malignant peripheral nerve sheath tumor (MPNST) is a malignant soft tissue sarcoma with high mortality, low morbidity, and poor prognosis. The MPNST occurs mostly in the limbs and torso, and rarely in the head and neck. However, MPNST is insensitive to radiotherapy and chemotherapy, and complete surgical resection with negative margin is the most important and effective strategy. We present a case of MPNST in the head and neck. The tumor invades the left temporal bone, petrous bone, and mastoid bone, and compression changes in the focal cerebellum and sigmoid sinus. The patient underwent the left temporal region tumor resection + surgical reconstruction with temporalis muscle flap and pectoralis major myocutaneous flap. Adjuvant radiotherapy (55 Gy) was given after surgery, and there were no local recurrence and distant metastasis after 31-month follow-up.
Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a type of malignant tumor originating from peripheral nerve or nerve sheath cells but does not include tumors originating from the epicardium and peripheral neurovasculature. In 2002, World Health Organization (WHO) classified neurosarcoma, neurofibrosarcoma, malignant schwannoma, and malignant neurilemmoma into MPNST. And in 2013, WHO classified MPNST into soft tissue tumors, including malignant peripheral epithelioid nerve sheath tumor and malignant triton tumor, belonging to class III to IV of nervous system tumor classification. Because of the low incidence in the population, there is no unified and standardized strategies for MPNST in clinic, and the treatment principle of soft tissue sarcoma is often referred to. Malignant peripheral nerve sheath tumor usually occurs in human bodies’ trunk and limbs, and rarely in the head and neck regions. And there were few articles about such MPNST with large volume and complete envelope. So we presented a case of MPNST that located in the region of head and neck and a literature review.
Case Presentation
A 55-year-old Chinese man came with a left head and neck mass that recurred for 3 years and the surface skin ulcer for 1 month. He had 2 operations and postoperative radiotherapy in past medical history. Eight years ago, the patient underwent a simple left head and neck mass resection in the local hospital, and the postoperative pathology was myofibroblastoma. Unfortunately, the tumor recurred 5 years ago and was surgically removed again. The pathology was neurilemmoma, and reversible focal cells proliferated actively. So postoperative radiation therapy was performed (3 times but unknown dose). Three years ago, the tumor relapsed again, gradually getting larger, with a tougher quality, fixed location, and pain (Figure 1). Imaging examination showed an 8.3 × 7.2 × 11.0 cm3 cauliflower-like mass on the left head and neck region, with bone destruction of the left temporal, and compression changes in the cerebellum and sigmoid sinus (Figures 2 and 3). Based on these findings, we initially suspected that the tumor is a head and neck MPNST.
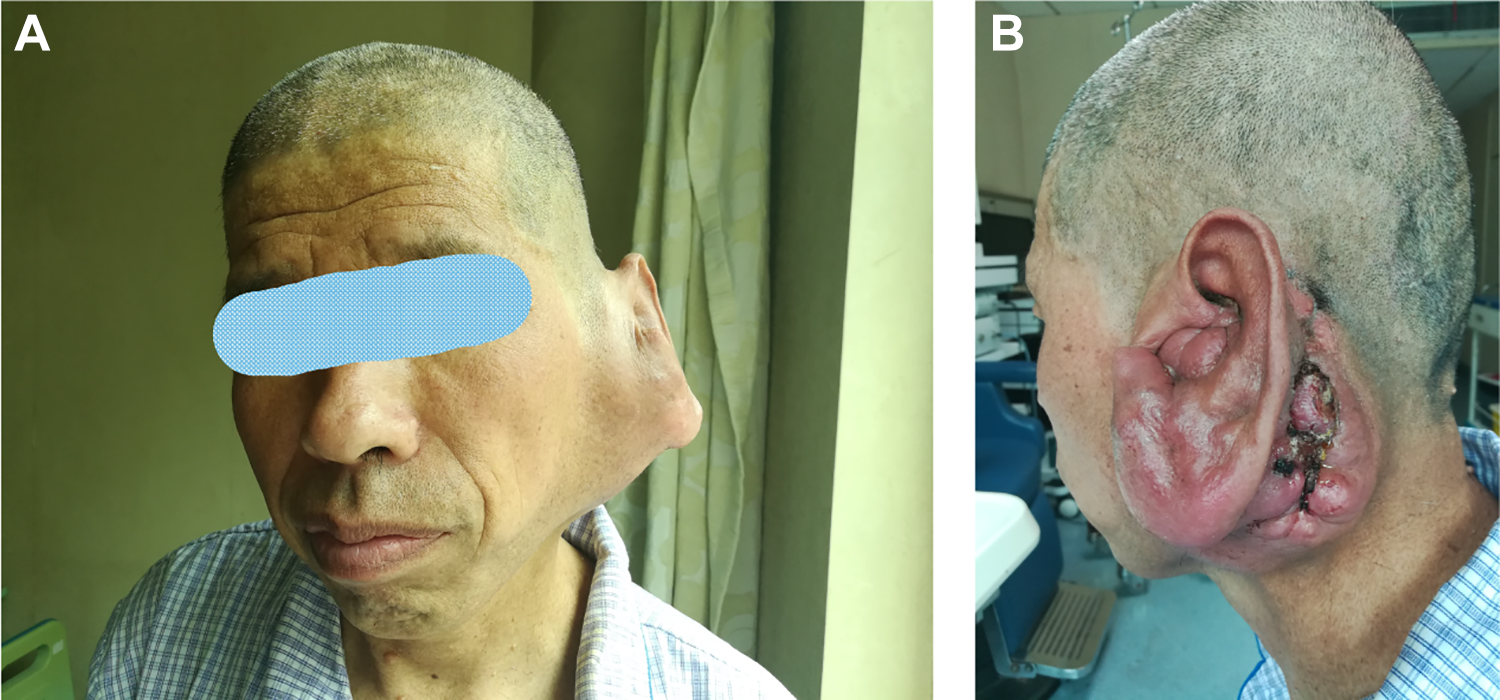
A, Front view; (B) side view. The patient’s surface skin broken and external auditory canal was clogged by the tumor.
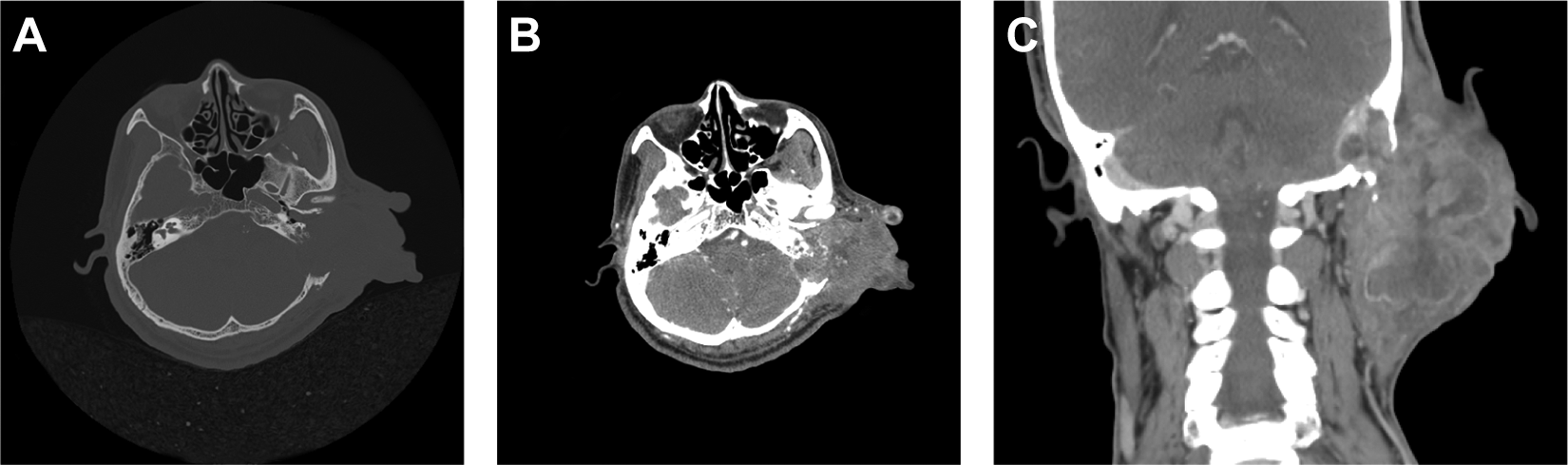
The preoperative CT scans (plain + enhanced) showed a huge cauliflower figure on the left side of temporal and neck region. A, Plain CT scan showed the tumor eroded the temporal bone, and the normal anatomical structure of the left middle ear cavity had disappeared. B, The tumor more reliably enhance after contrast administration, and had a clear boundary with the brain tissue. C, In coronal position, the tumor was huge and its size reached 11.0 cm (longitudinally) × 7.2 cm (laterally), and the tumor diffuse inhomogeneous enhancement after contrast administration.
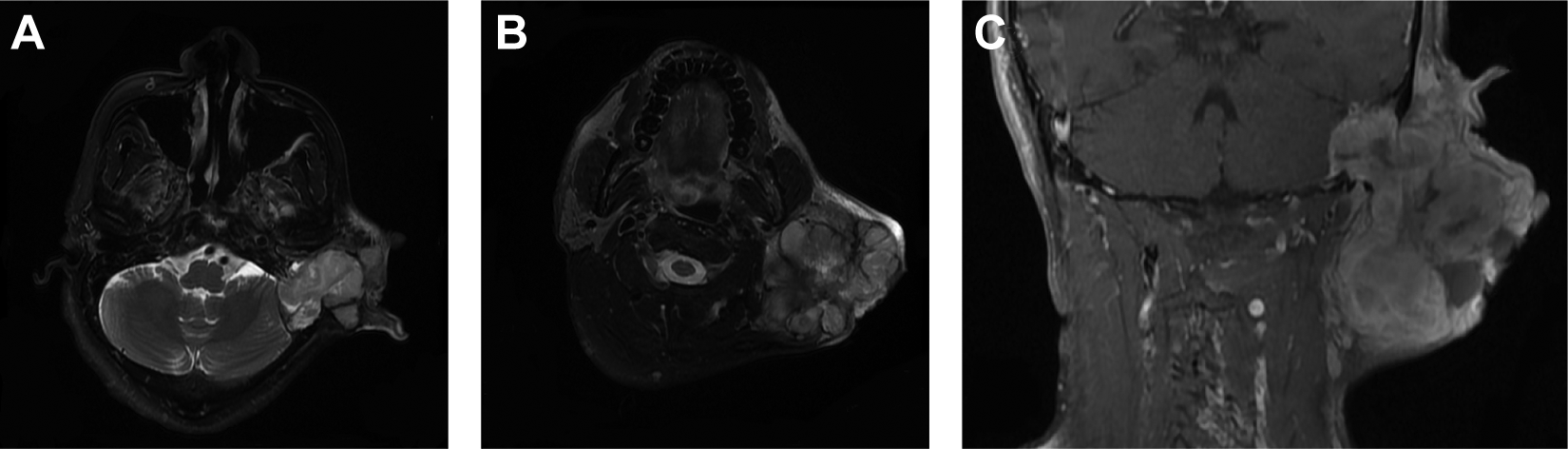
The preoperative MRI showed the tumor, on the left side of temporal and neck region, broke through into the brain, and the dura mater and sigmoid sinus were under pressure, and the dura mater was intact. A, T2-weighted horizontal images showed the left cerebellar hemisphere was compressed, and clearly demarcated. B, On T2-weighted horizontal images, the tumor appeared to be nodular or lobular. C, T2-weighted coronal images showed the tumor was giant with intracranial invasion.
After multidisciplinary discussion, the left head and neck region tumor resection + one-stage repair of the tissue defects with temporalis muscle flap and pectoralis major muscle flap. Intraoperative findings showed that the tumor was rich in blood supply, its capsule was intact, and it adhered tightly to the posterior margin of the parotid gland. Simultaneously, the tumor invaded the skull, and the middle ear was eroded by the tumor and loses its normal structure, and the dura mater was intact with no cerebrospinal fluid leakage occurring. Finally, the tumor was completely removed with negative intraoperative margin, and the tumor size was about 10 × 8.5 × 7 cm. And freeing the temporalis muscle flap and pectoralis major muscle flap, then using the free flaps to fill and reconstruct the temporal large defects. (Figure 4). Histopathology was an MPNST, involving temporal muscle and mastoid. Immunohistochemical staining showed that the tumor cell was all positive for S100, smooth muscle actin (SMA), Vimentin, and partly positive for CD34 antigen (CD34) and tumor protein 53 (P53), while negative for CD99 antigen, cytokeratin, Desmin, epithelial membrane antigen, human macrophage metalloelastase 45, neurofibromatosis (NF), and antigen Ki67 (Ki-67) index >20%.
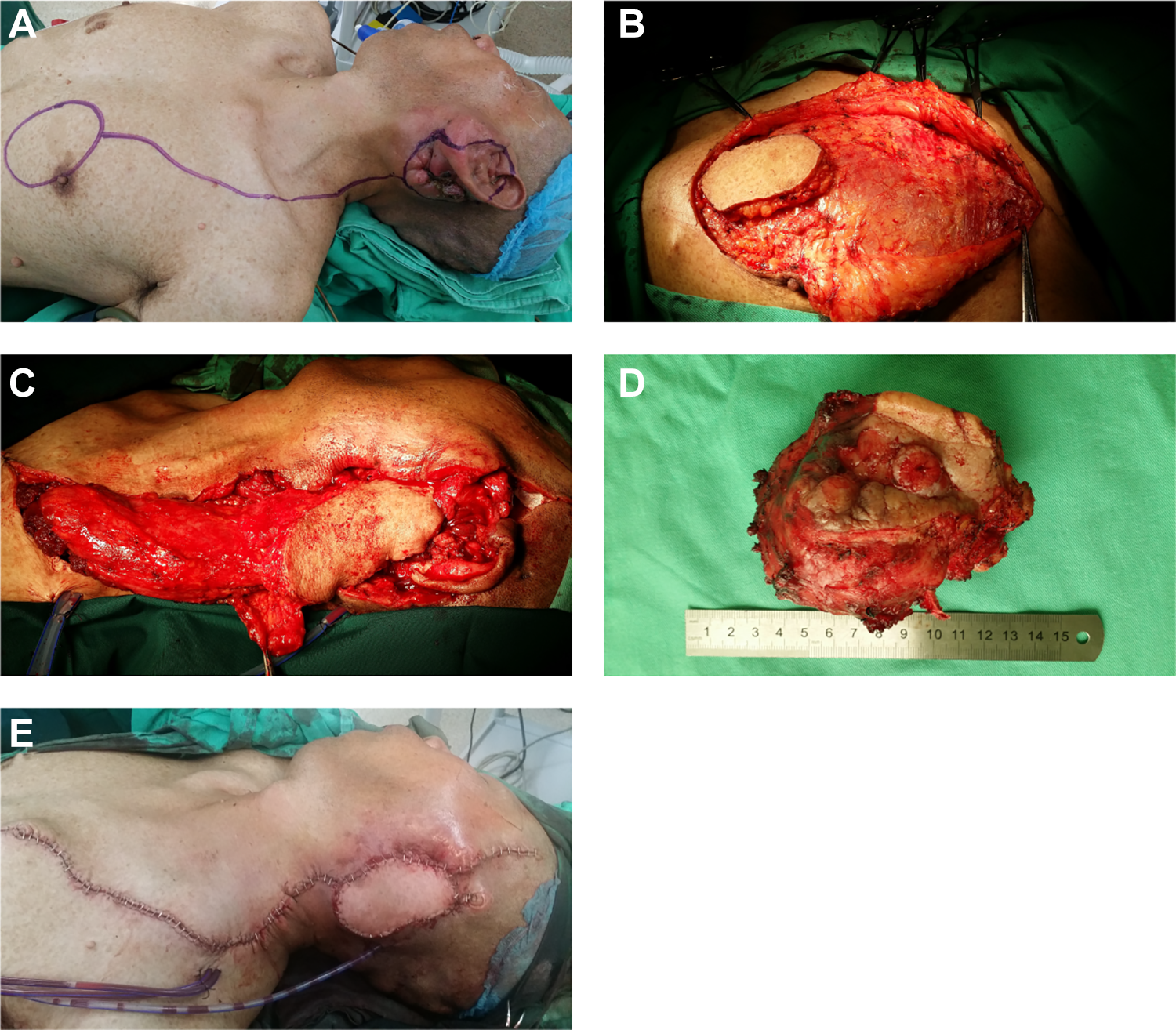
A, Preoperative surgical incision design; (B and C) free pedicled pectoralis major myocutaneous flap; (D) the mass was completely resected, and its capsule was intact; (E) reconstruct the defect through free myocutaneous flaps.
The incisions were healed by first intention, and no facial paralysis happened. One month later, the adjuvant radiotherapy patient was performed in the local hospital, and the dose was 55 Gy. There was no local recurrence or distant metastasis in the tumor after 31 months of follow-up.
Discussion
Malignant peripheral nerve sheath tumor is a rare malignant soft tissue tumor with neural differentiation potential, accounting for 5% to 10% of all soft tissue sarcomas. 1,2 The incidence of MPNST is 0.001%, and about 40% to 50% is secondary to malignant transformation of neurofibromatosis type 1 (NF1), and the rest is secondary to other tumors after radiotherapy and sporadic MPNST cases. Sporadic MPNST usually occurs in the elderly population, and radiotherapy-related MPNST occurs mostly in patients with breast cancer or lymphoma after receiving radiotherapy (median latency: 15-16 years). 3,4 A study based on the surveillance, epidemiology, and end results database found that the mean age of patients diagnosed with primary MPNST was 50.7 ± 22.4. 3 About 8% to 13% of NF1 develops into MPNST, and patients with internal plexiform neurofibromatosis have a higher risk of onset. 5 -7 At the same time, patients with NF1 had an earlier onset age of MPNST compared with non-NF1 patients (median age: 28 vs 41). 8
So far, the pathogenesis of MPNST has not been clarified. The NF1-MPNST is mainly related to NF1 gene mutation, and the changes in X-linked alpha thalassemia/mental retardation syndrome (ATRX), polycomb repressive complex 2 (PRC2), MET- c-MET, gene of phosphate and tension homology deleted on chromsome 10 (PTEN), insulin-like growth factor 1 receptor (IGF1R), epidermal growth factor receptor (EGFR), mitogen-activated protein kinase (MAPK), and other levels are also related to the progression of MPNST. 1,9 PTEN, IGF1R, and EGFR disrupt the normal cell cycle by affecting the phosphoinositide 3-kinase/protein Kinase B/mammalian target of rapamycin pathway, while MAPK by affecting the rapidly accelerated fibrosarcoma/mitogen activated extracellular signal regulated kinase/extracellular regulated protein kinases pathway. And the changes in ATRX and PRC2 expression levels are mainly related to tumor malignancy in NF1 patients.
Malignant peripheral nerve sheath tumor has a high degree of malignancy and is highly invasive. The clinical features are rapid growth of the mass, accompanied by pain, local compression, and motor and sensory nerve dysfunctions. Malignant peripheral nerve sheath tumor occurs mostly in the trunk and limbs, and a few occur in the head and neck. 10,11 In the study by Fan et al, 12 statistical analysis of tumor location in 146 Chinese patients with MPNST showed that 22.6% (33/146) were located in the head and neck, 43.8% (64/146) were located in the trunk, and 33.6% (49/146) were located in the extremities. It is basically consistent with the statistical results of Stucky et al. 10
Although computerized tomography, magnetic resonance image scanning, and fluorodeoxyglucose positron emission tomography have certain auxiliary effects on the differential diagnosis of MPNST, it still needs to rely on histopathology and immunohistochemistry to confirm. 13 Typical pathological features are that spindle cells are arranged in bundles, and some tumor areas show large necrosis and increased perivascular cells. And immunohistochemical staining is a vital method for identifying MPNST from other tumors. Tumor immunohistochemical staining markers S100, Ki-67, neuron-specific enolase, P53, SMA, Vimentin, CD34, and H3K27me2 deletions are good diagnostic indicators of MPNST. 14,15 A study found that SOX10, neurofibromin, and p16 expression are completely absent and EGFR positive is highly suggestive of MPNST. 16
Because of poor prognosis and insensitive to radiotherapy and chemotherapy, complete surgical resection of the tumor and negative margin is the main treatment of MPNST. The rate of local recurrence and distant metastasis is very high, and distant metastasis mainly shifts to the lungs. The clinical characters of MPNST indicating poor prognosis include tumor size >5 cm, high tumor grade, NF1 correlation, distant metastasis, surgical margin positive, S100 negative, and Ki-67 index >20%. Multiple studies have shown that the 5-year overall survival rate of MPNST is 44% to 57%, the 5-year disease-specific survival rate is 32% to 50%, and the prognosis of head and neck MPNST is worse. 12,16 -19 Watson et al 20 found that radiotherapy-associated MPNST had the worst 5-year disease-specific survival rate compared with NF1 and sporadic MPNST (47% < 52% < 67%). Therefore, for different types of MPNST, based on the complete surgical removal of the tumor, individualized treatment should be used.
In this patient, the postoperative pathological were myofibroblastoma and neurilemmoma with focal cells proliferating actively, respectively. And unknown dose of adjuvant radiotherapy after the second surgery. Therefore, it is hard to define whether the MPNST is derived from a malignant transformation of neurilemmoma or secondary to the unknown dose of radiation therapy. And the tumor volume was large, the surrounding bone was invaded, and a huge defect after tumor resection. Companied with the positive of S100 and Ki-67 index >20% indicates a poor prognosis. Fortunately, the tumor had an intact envelope and was completely removed with negative margin. Adjuvant radiotherapy (55 Gy) was given after surgery. And there were no recurrences and metastases after 31-month follow-up.
In conclusion, surgical resection completely with a negative margin is still the main treatment for MPNST, and postoperative adjuvant radiotherapy to improve prognosis. The head and neck MPNST often invade important tissues and organs and facing defects after tumor resection. So, the free myocutaneous flaps (such as pectoralis major myocutaneous flap, temporalis muscle flap, clavicular myocutaneous flap, etc) are inevitably needed for reconstruction in head and neck.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.