
Abstract
Ti-based alloys have the potential to be used as hydrogen storage units, including NiTi. In contrast, NiTi alloy is sensitive to H atoms. It has been found that hydrogen can cause embrittlement in NiTi alloys. Thus, it is become indispensable to elucidate the reaction of H2 molecules on the NiTi surface. Using density functional theory, we investigated the dissociation mechanism of H2 molecules on the B2 NiTi (001) surface. We found that H atoms tend to come closer to Ni atoms on the Ti- and Ni-terminated (NiTi) (001) substrate. The calculation results showed that the adsorption energy of H atoms at the hollow site was higher than that at the top site. We identified two dissociation mechanisms of H2 molecules on Ti and Ni terminated on NiTi (001) substrates via the hollow sites of the adsorption route. The adsorption energy values obtained were extremely low, that is, 0.23 and 0.38 eV for the Ni and Ti terminated of NiTi (001) substrates, respectively. The dissociation reaction of H2 molecules, which is an exothermic reaction, can quickly occur on the NiTi (001) surface because of the low activation energy.
Introduction
NiTi alloys are materials that can return to their original shape after undergoing deformation and are therefore called shape memory alloys. 1 The properties of such alloys are related to the thermo-mechanical ability of NiTi alloys to transform from the martensite-austenite phase, and vice versa.2,3 These alloys are widely applied in various engineering fields, including aerospace,4–6 automotive7,8 and civil engineering.9,10 Cables 11 and double layers of NiTi films 12 have been proven to dissipate stress. NiTi alloys are widely used in the medical field as orthodontic arch-wires, bone implants, and heart stents.1,3 Theoretical study on the atomic diffusion at the NiTi liquid interface has been reported recently. 13 In that study, it has been reported that the diffusion activation energy of Ni is 0.2 eV higher than that of Ti. 13 In other studies, it has been reported that there are differences in the structure of the glassy NiTi formed through several variations of cooling time. 14
Ti-based metal alloys are known to be sensitive to H atoms. 15 The H atoms in metals tend to occupy interstitial locations and therefore cause distortions of the host lattice. The presence of H atoms can change the properties of the original metal alloy. 15 The results of a recent study showed that hydrogen could cause embrittlement in NiTi alloys after several loading cycles. 16 In contrast, Ti-based alloys can be used as hydrogen storage units, including NiTi. 17 Although several previous theoretical studies have been performed to investigate hydrogen absorption on NiTi, 17 the adsorption of dissociated H atoms from CH4 molecules on the Ni(111) surface, 18 and H defects in NiTi alloys. 19 In addition, several mechanisms of the dissociation reaction processes of hydrocarbon molecules have been reported in the previous studies within the framework of DFT.20–22 Shibuta et al. have investigated the dissociation of ethanol on the surface of Pt. 20 Likewise, the mechanism of dissociation of ethylene molecules on the Ni cluster and on the Ni (111) surface has been reported by Shimamura et al. 21 and Arifin et al., 22 respectively. However, to the best of our knowledge, no study has yet investigated the dissociation mechanism of H2 molecules on the NiTi surface. Therefore, in this study, we calculated the activation energy to explain the mechanism of the dissociation reaction of H2 molecules on the NiTi (001) surface using density functional theory (DFT) method. We aimed to demonstrate the H2 decomposition process on the NiTi (001) surface. The results of this study are expected to provide preliminary information about how H2 molecules are adsorbed on the NiTi surface. In the future, this information will be useful in two different areas of technology, namely in the influence of H atoms on the mechanical properties of NiTi alloys, and in the potential of NiTi alloys as hydrogen storage units.
Numerical methods
All electronic calculations were performed using Quantum Espresso 23 within the DFT framework. The Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation was performed for the exchange correlation energy. Pseudopotentials for Ni, Ti, and H were obtained from the SSSP precision library provided on materialscloud.org. 24 Norm-conserving pseudopotential 25 is used for H atoms, and ultrasoft pseudopotentials 26 are implemented for Ni and Ti atoms. The lattice parameter of the NiTi alloy was calculated to be 3.01 A, which agrees with the value reported in a previous study. 27 The plane-wave cutoff energies were 45 Ry and 360 Ry for the electronic pseudo-wave function and pseudo-charge density, respectively. Four layers of NiTi (001) containing nine atoms in each layer were used as the substrate, as shown in Figure 1. Figure 1(a) and (b) show the Ti alloy substrates terminated by Ni and Ti atoms, respectively. The atoms in the two bottommost layers were fixed to mimic the slab system. A vacuum space of 12 Å was introduced in the system. The Brillouin zone was sampled using a 2 × 2 × 1 Monkhorst–Pack scheme. To calculate the adsorption energy, the H atoms were initially placed 2.0 Å above the substrate at the top and hollow sites, and then structure optimization was carried out. The adsorption energy Eads was calculated using the formula Eads = (ENiTi + EA) –ENiTi/A, where ENiTi denotes the energy of the NiTi (001) substrate, EA denotes the energy of the isolated adsorbate, and ENiTi/A denotes the energy of the system containing the substrate and adsorbate. To investigate the type of adsorption of H2 molecules on the NiTi surface, H2 molecule was placed 2.0 Å above the NiTi surface at the top and hollow sites, and the system was minimized. The nudged-elastic-band (NEB) method was employed to search the minimum energy path (MEP) and to estimate the activation energy of the H2 dissociation reaction on the NiTi (001) substrate. In our calculation, the criterion for the convergence of the MEPs was 0.05 eV/Å. Initially, the reactant configurations and product configurations are interpolated linearly to find some intermediate configuration. A total of 10 configurations, hereinafter referred to as replicas, were used in this study. Furthermore, the replicas connected to the elastic spring are optimized toward the minimum energy path. The activation energy of a reaction is the energy difference between the energy of the transition state and the energy of the reactants. The atomic configurations were visualized using the XCrySDen visualization program. 28
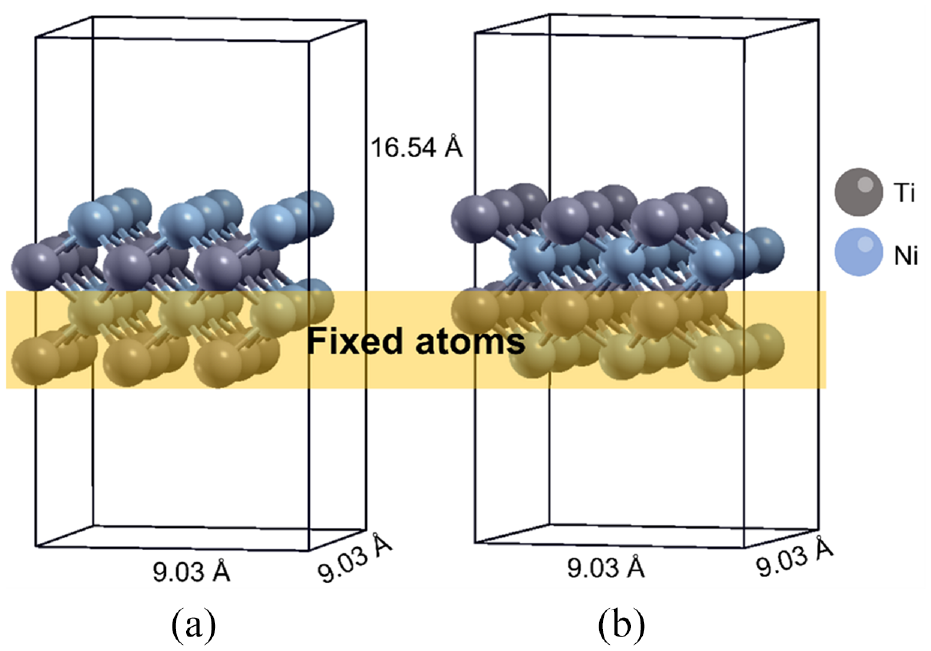
Initial structure of NiTi (001) substrate terminated by Ni and Ti atoms for (a and b), respectively. Blue and gray balls correspond to the Ni and Ti atoms, respectively. The two layers of atoms in the bottom, blocked by transparent yellow color, are fixed during the optimization.
Results and discussion
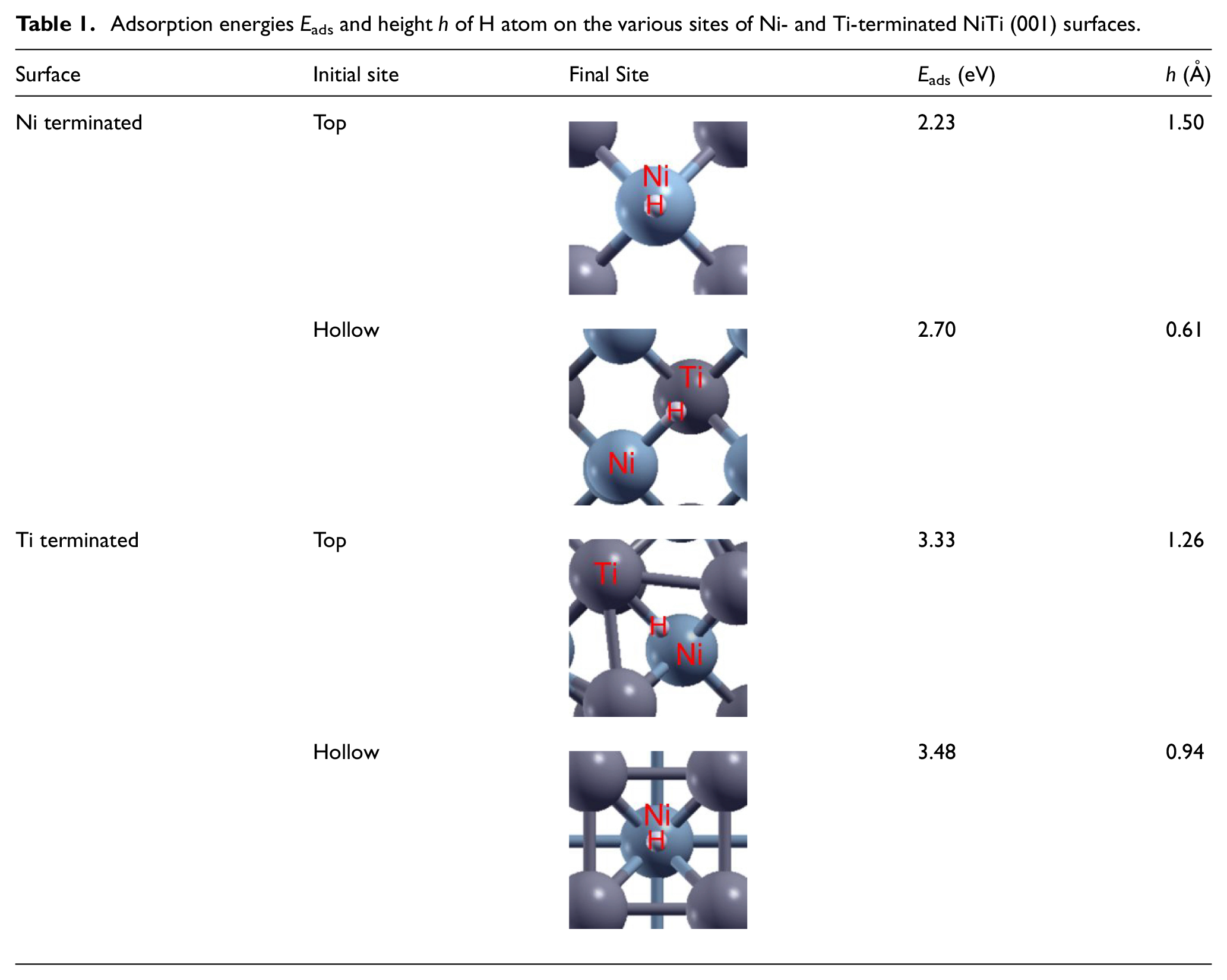
In this technical note, we examined the adsorption of H atoms on the NiTi (001) surface at top and hollow sites. Table 1 lists the location of the H atoms on the NiTi (001) surface after optimization, the adsorption energy value, and the distance of the H atoms from the surface. As can be seen, after optimization, the H atoms that are directly above the Ni atoms, at the top sites on the Ni-terminated surface and the hollow sites on the Ti-terminated surfaces, remain at these sites. The H atom above the Ti atom shifts toward the Ni atom on the NiTi surface with the difference of 0.47 and 0.18 eV on the Ni and Ti terminated surfaces, respectively. Notably, hollow sites are preferred by the H atoms compared to top sites and the adsorption energy of H atoms at the hollow site on the NiTi surface is higher than that at the top site. The simulation results show that the H atoms are closer above the NiTi (001) surface at the hollow site than at the top site. Additionally, the adsorption energy on the Ti-terminated surface was higher than that on the Ni-terminated surface for both the top and hollow sites. These results are consistent with those presented by previous studies, which reported the preference of adsorption of H atoms on the Ti-terminated surface in several types of binary alloys. 29 It is interesting to note that from the results of the optimization of the NiTi substrate structure, the energy in the Ti terminated system is less negative than in the Ni terminated system with a difference of 0.86 eV in our calculations; this contributed to the adsorption energy values.
Adsorption energies Eads and height h of H atom on the various sites of Ni- and Ti-terminated NiTi (001) surfaces.
Furthermore, we calculated the activation energy for the dissociation of H2 molecules on the NiTi (001) surface. To observe possible H2 dissociation routes on the NiTi (001) surface, we placed H2 molecules at the top and hollow sites on the NiTi (001) surface and then performed structural optimization. We found that the H2 molecules placed in the hollow sites were chemically adsorbed on the surface from the optimization results. The optimization results on the Ti-terminated surface show that the H2 molecule dissociates into two H atoms, where one H atom was on the surface while the other H atom was on the subsurface. We set the optimization results as the initial and final state configurations on the Ni (see Figure 2(a)) and Ti (see Figure 2(b)) terminated surfaces, respectively. Using the NEB method, we estimated the activation energy of the dissociation reaction of H2 molecule on the terminated Ni and Ti surfaces to be 0.23 and 0.38 eV, respectively. These values are significantly lower than the dissociation energy of pure H2 molecules, which is 4.52 eV. Thus, the NiTi (001) substrate can also act as a catalyst in the dissociation of H2 molecules which can reduce the activation energies by 4.29–4.14 eV for terminated Ni and Ti, respectively. Both the reactions in our calculation were highly exothermic at 0.83 and 0.5 eV for Ni and Ti terminated surfaces, respectively. It can be seen in Figure 2(a) and (b), that the energies of the products are much lower than the initial energies before the dissociation process occurs. In this case, a certain amount of energy is released because of the dissociation reaction. These results indicated that both the reactions as mentioned above occurred spontaneously; however, this was not the case with a reverse reaction, that is, when an H2 molecule was formed from two H atoms. This is because such reactions require a considerable amount of formation energy to occur. Furthermore, future studies are needed to investigate the H2 dissociation on other NiTi surfaces. The results of this study can motivate further research on how to prevent the diffusion of H atoms in NiTi alloys causing distortions in the host lattice. On the other hand, further research on the potential of NiTi as a hydrogen storage unit will also be a challenge.
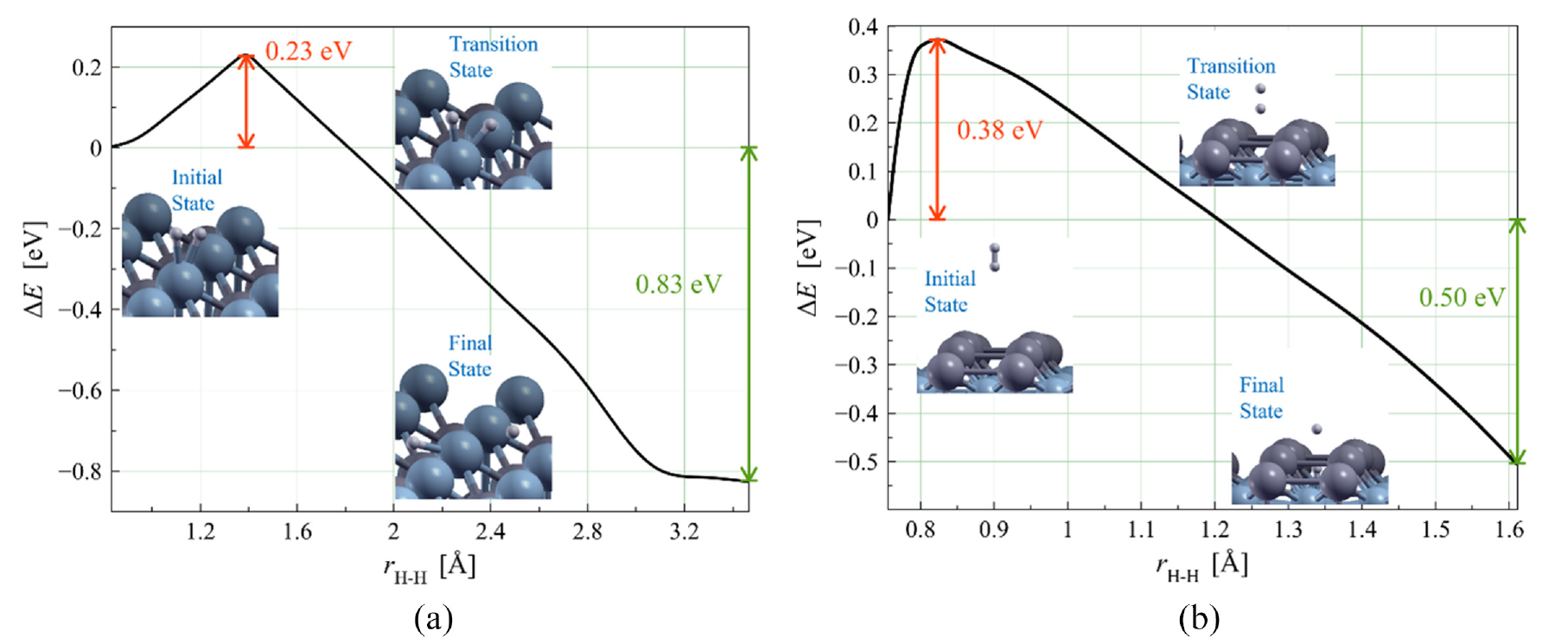
Activation energy plot and their related structure of the two dissociation mechanisms of H2 molecules on the NiTi (001) substrate: (a) the H2 dissociation on the Ni terminated NiTi alloy and (b) the H2 dissociation on the Ti terminated NiTi alloy.
Conclusions
It can be concluded from our results that the H atoms adsorbed on the NiTi (001) surface move closer to the Ni atoms on the surface. We also found that the hollow sites are preferred by H atoms on the NiTi (001) surface compared to the top sites; this is indicated by the higher adsorption energy at the hollow sites. The NiTi substrate (001) also acts as a catalyst in the dissociation reaction of H2 molecules, which can significantly reduce the dissociation energy from 4.14 to 4.29 eV. The dissociation reaction of H2 molecules, which is an exothermic reaction, can quickly occur on the NiTi (001) surface because of the low activation energy. Further investigations need to be performed for other NiTi surface planes. The results of this study can be used as a reference for further research to find ways to prevent the infiltration of H atoms in NiTi alloys that cause embrittlement of the material. In addition, these results can also motivate further research on the potential of NiTi as a hydrogen storage unit, which is still a challenge until now.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a PDUPT 2021 research grant from the Ministry of Education, Culture, Research, and Technology of the Republic of Indonesia [contract # 167/E4.1/AK.04.PT/2021], and the manuscript preparation was done in a World Class Professor Program 2021 [# 2817/E4.1/KK.04.05/2021].