
Abstract
Objective
To describe the redactions in contemporary protocols for industry-sponsored randomised drug trials with patient relevant outcomes and to evaluate whether there was a legitimate rationale for the redactions.
Design
Cohort study. Under the Freedom of Information Act, we requested access to trial protocols approved by a research ethics committee in Denmark from October 2012 to March 2013. We received 17 consecutive protocols, which had been redacted before we got them, and nine protocols without redactions. In five additional cases, the companies refused to let the committees give us access, and in three other cases, documents were missing.
Participants
Not applicable.
Setting
Not applicable.
Main outcome measure
Amount and nature of redactions in 22 predefined key protocol variables.
Results
The redactions were most widespread in those sections of the protocol where there is empirical evidence of substantial problems with the trustworthiness of published drug trials: data analysis, handling of missing data, detection and analysis of adverse events, definition of the outcomes, interim analyses and premature termination of the study, sponsor’s access to incoming data while the study is running, ownership to the data and investigators’ publication rights. The parts of the text that were redacted differed widely, both between companies and within the same company.
Conclusions
We could not identify any legitimate rationale for the redactions. The current mistrust in industry-sponsored drug trials can only change if the industry offers unconditional access to its trial protocols and other relevant documents and data.
Keywords
Introduction
Trial protocols are needed for a proper assessment of the veracity of drug trial reports, but it is difficult to get access to them.1,2 Drug companies routinely claim that they contain commercially sensitive information, trade secrets or intellectual property, although there seems to be no commercial secrets in trial protocols. 2
We experienced difficulties when we applied for access to a cohort of research protocols for ongoing drug trials recently approved by one of the five regional research ethics committees in Denmark under the Freedom of Information Act. We wished to compare the information in protocols with the information provided to the patients in order to evaluate whether the trials were ethical and necessary and whether essential information about the benefits and the harms of the drugs had been hidden from the patients. On several occasions, the regional committees allowed the sponsor to delete vast amounts of information important to our research, and in some cases, we did not even receive a protocol. After having appealed to the National Committee on Health Research Ethics, we received protocols for all the trials, which were often much less redacted than those we had received from one of the local committees. This gave us a unique opportunity to evaluate whether the many deletions made sense and were consistent across and within companies.
Methods
We used the website of the Danish National Committee on Health Research Ethics to screen all titles of studies approved by one of the five regional ethics committees in Denmark from January 2012 to March 2013 (1401 projects).
The information in the database is restricted to the date of approval, project title, regions where the study will be conducted and the name of the coordinating investigator. We searched the World Health Organization International Clinical Trials Registry Platform and clinicaltrials.gov using the study ID, or title or keywords in the title from the 1401 projects as search terms. We supplemented with Google if we were unable to find the study in the trial registries. The information collected included study type, design, population, interventions, inclusion and exclusion criteria, primary outcomes and estimated sample size.
We were only interested in randomised trials that could be directly relevant for patients and we therefore excluded studies using surrogates as primary outcomes, e.g. blood pressure and cholesterol, as they tend to overestimate the benefit of drugs and to overlook serious harms. 3 We also excluded trials with a cross-over or cluster randomised design, as we aimed at collecting a cohort of projects that were reasonably comparable in design. One observer screened the projects and consulted an additional observer when there was uncertainty about eligibility.
We found 212 projects that seemed to fulfil our criteria, and as we aimed for a minimum sample of 60 protocols, which our previous research has shown is sufficient for projects like the current one,4–7 we requested access to 78 trial protocols approved by a regional committee within the most recent six months (October 2012 to March 2013).
We asked the committees for full access to the protocol and any related documents, including those on publication rights, which are not always part of the protocol. We emphasised that our results would be published in a manner that did not allow identification of individual trials and referred to our previous research on protocols,4–7 which had led to important results for patient safety.
To evaluate systematically the characteristics of the redactions, we adapted a framework used in a Cochrane review comparing the content of protocols with that in trial reports. 8 Based on this review, we defined 22 key protocol variables. One observer (MM) went through the redacted protocols and noted if the sections corresponding to our variables had been partly or completely concealed. As this assessment was somewhat subjective, all ambiguous cases were discussed with a second observer (PCG). It was usually possible to infer what had been blackened out from the table of contents or from the unredacted text we had received after our appeal to the national research ethics committee. In some protocols, we could reveal what was hidden by copying the blackened out text and pasting it into a Word document. When it was impossible to tell if the sections we looked for had been redacted or were simply not mentioned in a protocol, we categorised them as unknown.
Results
It proved very difficult to obtain all 78 protocols we requested from the local ethics committees and the original unredacted protocols had been extensively redacted before we got them. The national research ethics committee offered us full access provided we signed a confidentiality agreement stating that we would not share information in the protocol with others, which a sponsor regarded as confidential. Despite this reassurance, several companies refused to provide their protocol and involved their lawyers. Sanofi-Aventis sued the national committee but lost the court case. Even after one and a half years, the processes for several of the protocols were still ongoing. Finally, in 2016, three years after we started our project, we received the last of the protocols we had requested.
We excluded eight protocols that did not meet our inclusion criteria: five did not have a clinically relevant primary outcome; one had a cross-over design; one was not randomised; and one was a duplicate protocol.
Sponsoring companies and the amount of redactions in their protocols.
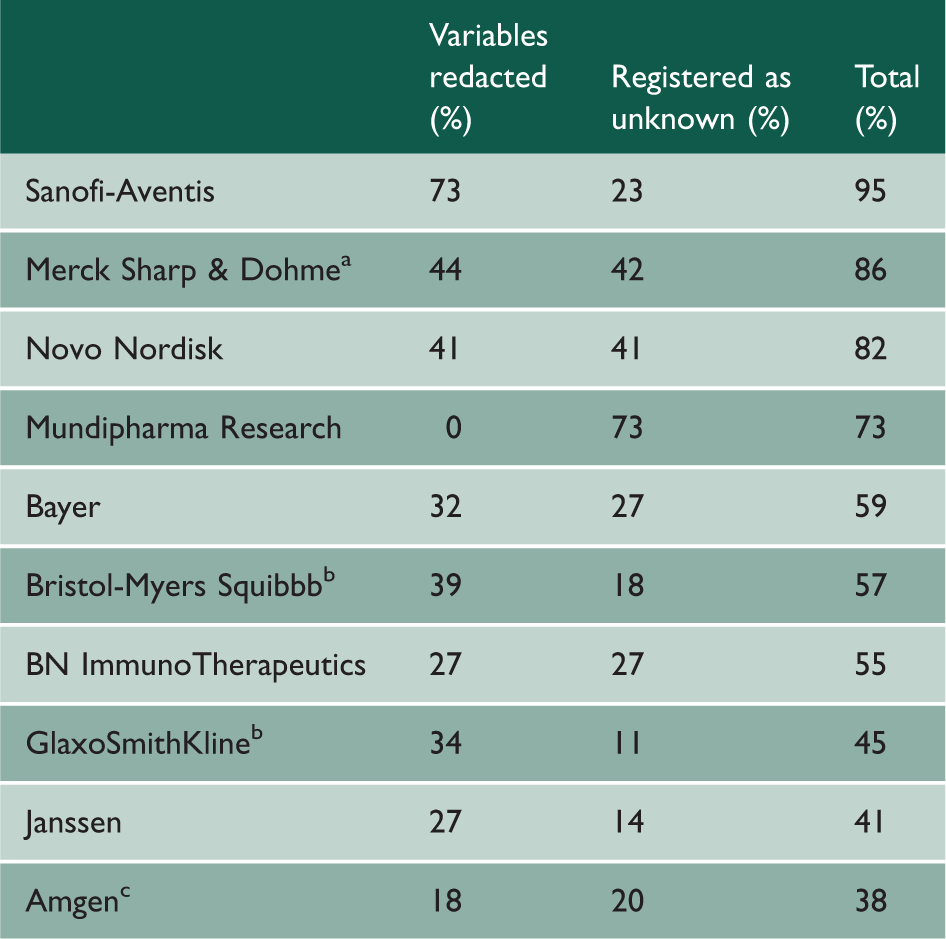
Average of afour protocols, btwo protocols and cthree protocols.
We describe here the deletions in the protocols for the 17 commercially sponsored trials that had redactions. The median number of pages in the protocols was 111 (range: 60 to 197). The extent to which our 22 variables had been redacted is presented in Figure 1, and we have ranked the companies according to their level of secretiveness in Table 1. We describe the most serious issues first.
Redacted information in 17 commercial protocols according to our 22 key variables.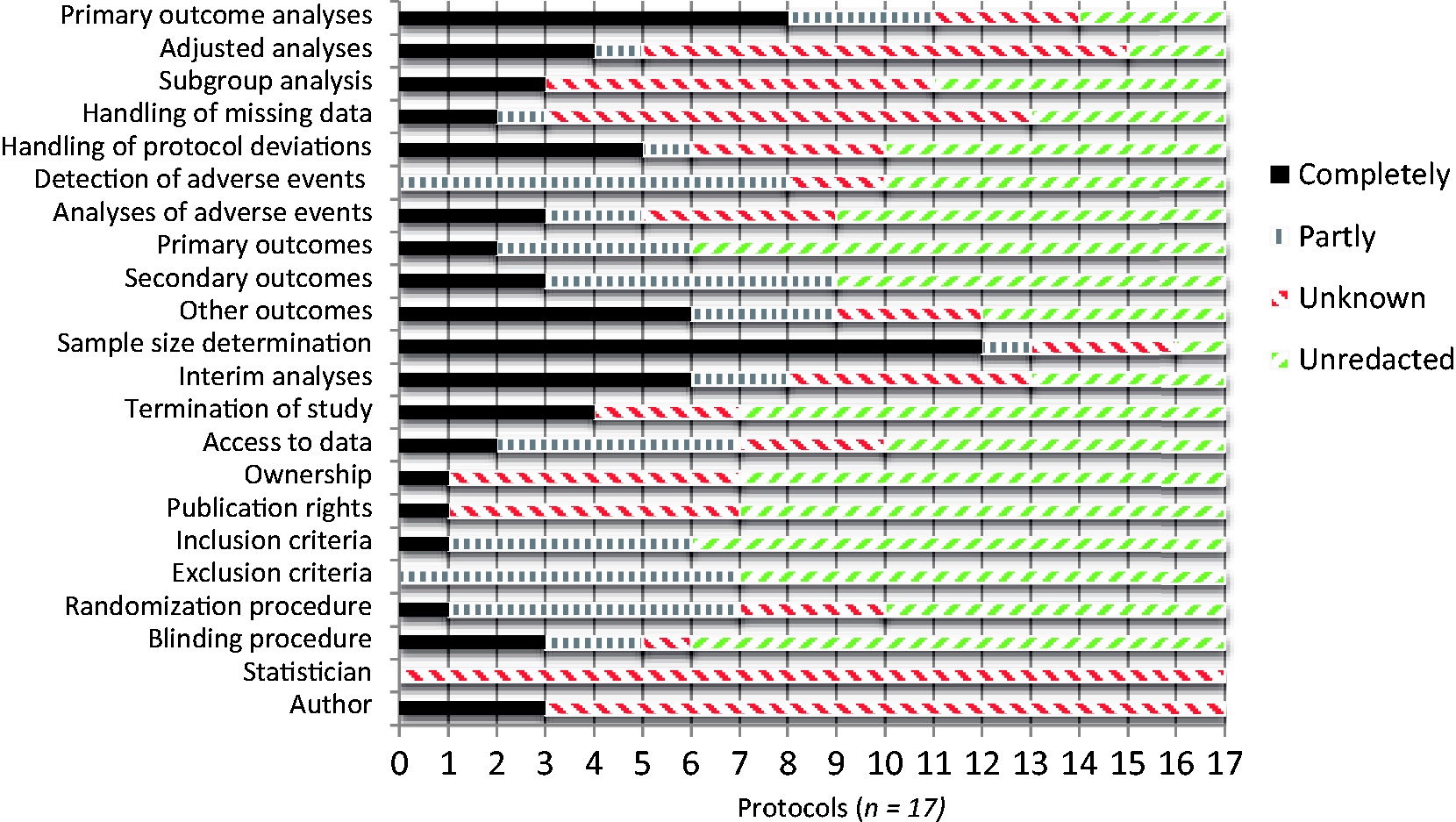
Primary outcomes
Everything about the analysis of the primary outcomes was redacted in eight protocols (47%). As for their definition, nothing was redacted in 11 protocols, while four had concealed parts of their primary outcomes (Sanofi-Aventis had redacted all six pages defining the five components of its composite primary efficacy endpoint). In the two remaining protocols, Bristol-Myers Squibb and GlaxoSmithKline had concealed all of the primary outcomes.
Secondary outcomes
In eight protocols, nothing was redacted while six had concealed parts of their secondary outcomes. In the three remaining protocols, Bristol-Myers Squibb, Merck Sharp & Dohme and Glaxo-SmithKline had concealed all of their secondary outcomes.
Other outcomes
In three protocols, we could not see what had been deleted. Partial redaction occurred in three other protocols, while in six protocols, everything was redacted in sections such as ‘Exploratory Outcomes’, ‘Other Endpoint(s)’ or ‘Other Efficacy Endpoint(s)’. In the remaining five protocols, there were no other outcomes.
Adjusted analyses
Only two companies had an adjusted analysis section we could read. In 12 protocols, everything was redacted in the relevant section on statistics, while in one, parts had been blackened out. We classified the remaining two protocols as unknown.
Subgroup analyses
Subgroup analyses were described in nine protocols, three of which had redacted the information completely, while the other six did not conceal them. We listed the remaining eight protocols as unknown.
Handling of missing data
Merck had redacted everything in two of its protocols; GlaxoSmithKline referred to a reporting and analysis plan we did not have, and Amgen had blackened out parts of the content. We classified ten protocols as unknown. In three protocols, nothing was redacted.
Handling of protocol deviations
In five protocols, everything was redacted, and in one, most of the text was gone. We classified four protocols as unknown. Seven protocols were not redacted, but one referred to a Standard Operating Manual we did not have.
Detection of adverse events
In two protocols by Merck and Mundipharma, it was not possible to assess how adverse events were to be handled, as the protocols were so heavily redacted that we registered them as unknown. Eight protocols were partly redacted and Sanofi-Aventis had redacted most of the text in the section ‘Obligation of the Investigator Regarding Safety Reporting’. Other companies also concealed essential information. In two of its protocols, Merck had redacted much of the information under ‘Safety Monitoring and Assessments’; under ‘Overdose’ all information of value was removed; and in one protocol, all information concerning ‘List of Safety Measurements’, ‘Selected Nonserious Adverse Experiences (if applicable)’ and ‘Definition of an Overdose for This Protocol’ was gone. Novo Nordisk had crossed out everything in the sections ‘Medical Events of Special Interest’ and ‘Precautions and/or Overdose’, while Amgen had blackened out the safety risks of its investigational drug. Seven protocols were not redacted.
Analyses of adverse events
In three protocols, everything was redacted. Two protocols were partly redacted, and in one of them, GlaxoSmithKline referred to a Reporting and Analysis Plan we did not have. Four protocols were registered as unknown; we could not assess whether safety analyses were described in the section on statistical considerations, as this was completely redacted. Eight protocols were not redacted.
Sample size determination
In 12 protocols, everything was redacted, and in one, some was redacted, which included the power and the assumed response rate. We classified three protocols as unknown. Only one protocol, from GlaxoSmithKline, had no redactions.
Interim analyses and termination of study
In six protocols, everything about interim analyses was redacted, and in two, parts were redacted. Five other protocols were registered as unknown. In two studies, there was no planned interim analysis, and in the remaining two protocols, we could read the section on interim analysis.
The termination criteria were completely redacted in four instances while three were registered as unknown. In one of these, Novo Nordisk had a readable section but referred to a data and safety monitoring charter for termination criteria, which we did not have. In the ten remaining protocols, the termination criteria were not concealed, but in nine, the sponsor reserved the right to terminate the study at any time. Merck was the only company with a protocol that had criteria for when the study could be ended prematurely but it sponsored another trial where it reserved the right to terminate the study at any time.
Access to the data during study conduct
In two protocols, Merck had completely redacted the sections on safety and monitoring committees, and there were redactions in five additional protocols. Seven protocols were not redacted and the remaining three were registered as unknown.
Furthermore, 11 protocols – six of which were registered as unredacted – referred to review charters we did not have access to. Thus, in most cases, it was unknown whether the sponsor could get access to the accumulating data during trial conduct.
Ownership to the data and publication rights
Sanofi-Aventis had crossed out all information on ownership. Six protocols had no redactions but a statement that the data collected during the study were the sole property of the sponsor. We could not find any information about ownership in the remaining ten protocols.
In one protocol, Merck had concealed all information about investigators’ right to publish, and Amgen had redacted around 80% of the text in a contract between the sponsor and investigator. Six of ten protocols without redactions had a statement that the sponsor needed to approve the manuscripts or could demand deletion of information considered confidential; in the other four protocols, the sponsor had the right to postpone the publication. Five protocols were registered as unknown; three of these referred to a charter or contract we did not have.
Eligibility criteria
Six protocols had redactions in the inclusion criteria and seven in the exclusion criteria. Sanofi-Aventis had redacted all inclusion criteria in its protocol and 28 of 30 exclusion criteria.
Randomisation procedure
Sanofi-Aventis had blackened out everything. Six protocols were partly redacted, which made it impossible to assess whether the randomisation procedure was adequate. Three additional protocols described the study as randomised, but we could not find a section with the procedure, and three were unredacted but referred to other documents for details, which we did not have access to. Thus, only four protocols revealed the randomisation procedure.
Blinding procedure
The blinding procedure was completely redacted in three protocols and almost completely redacted in two. We classified one protocol as unknown. The remaining 11 protocols were unredacted.
Authors and statisticians
None of the 17 protocols specified who the statistician was. Only three protocols had a section mentioning the author of the protocol but it was blackened out. In five protocols, the sponsor had removed the names of the study director, coordinating investigator, trial director or key sponsor contact person.
Discussion
To our knowledge, our study is the first systematic assessment of which information in trial protocols pharmaceutical companies do not wish to disclose to independent researchers. There is a case report whose author, a former member of a research ethics committee, was asked to participate in a study of the varicella zoster vaccine to prevent shingles. 9 He describes his difficulties in gaining access to the research protocol and gives an overview of the redactions when he finally received it. He sarcastically concluded that if the policy of withholding protocols from the public continued, the most interesting question was whether research ethics committees ought to require that participants should routinely be told of their right to access to redacted protocols.
The amount of redactions in the protocols we received was so vast that it made them rather useless for research purposes, e.g. for assessing the ethical justification for the studies and to identify discrepancies with subsequent publications. 6 If there had been a legitimate ethical rationale for the redactions, we would have expected them to be reasonably uniform across companies and within the same company, but this was far from the case. For example, Merck Sharp & Dohme redacted virtually everything in one of their protocols and much less in their other three protocols so that, on average, 44% of the variables had been redacted. Eli Lilly denied us access to one of their protocols but accepted we got another one without any redactions, and Bristol Myers Squibb denied us access to one protocol and redacted 39% of the variables on average in two other protocols.
The redactions were most widespread in those sections of the protocol where there is empirical evidence of substantial problems with the trustworthiness of published drug trials. There are no good reasons for redacting the definition and analysis of the primary outcomes, but there are unsound ones like hiding scientific misconduct. In the first systematic review comparing protocols with published trial reports, we found that at least one primary outcome had been changed, introduced or omitted in two-thirds of the reports, and this change was not mentioned in a single published report. 4
There are many ways in which one can handle missing data and make adjusted analyses, and it is concerning that only three and two protocols, respectively, had unredacted information about this. We have previously reported unacknowledged discrepancies between protocols and publications in 39 of 49 trials for missing data and in 23 of 28 trials for adjusted analyses. 5
It is also noteworthy that only one protocol had no redactions in the section on sample size determination, as this section is often changed post hoc to fit better with what was obtained. In our previous research, we found unacknowledged discrepancies for sample size calculations in 18 of 34 trials. 5
It was unclear in most cases whether the sponsor could get access to the data as they rolled in and everything about interim analyses was redacted in six protocols. Furthermore, in most cases, the sponsor could terminate the trial at any time. We previously found that the sponsor had potential control over a trial in progress in 32 of 44 trials and that the sponsor had access to accumulating data during 16 trials, e.g. through interim analyses and participation in data and safety monitoring committees, but such access was disclosed in only one corresponding trial report. 6 The possibility of inappropriate sponsor control over a trial in progress involves the risk that the trial is stopped when the data happen to be favourable for the sponsor. A systematic review of 143 trials stopped early for benefit showed that trials with fewer events yielded greater treatment effects (odds ratio: 28; 95% confidence interval: 11–73). 10
We have no idea why only four protocols revealed the randomisation procedure, as we cannot imagine any commercial advantage in hiding it.
There was much secrecy related to the detection and analysis of adverse events, another area where published trial reports are often unreliable and often underestimate seriously the harms of drugs even to the point of omitting most or all of the deaths that occurred.11,12
Finally, it is troubling that there was so little information about ownership to the data and publication rights in the protocols. When such information is published, it is often unreliable. We got access to the full, unredacted protocols in a cohort of industry-sponsored trials published in The Lancet and found that there was no information on academic authors’ access to the data in 67 of the 69 protocols. 13 This was in striking contrast to the published papers, which indicated that one or more academic authors had access to the data in 64 trials. It seems to have become more common in recent times that publication agreements appear in a document separate from the protocol, which no one has access to, apart from the company and its academic investigators. 6
Limitations
We did not address all the redactions in the protocols, as we focused on a set of predefined variables, and we did not have access to supplementary documents that the companies had not submitted to the research ethics committees.
Conclusions
Reproducibility, and meticulous checking of the results and the risk of bias, also by people not involved with the research, and comparing what was published with what was planned, are essential elements in science. When this is not possible, science ceases to exist. 14 The current mistrust in industry-sponsored drug trials can only change if the industry becomes collaborative about everything that matters for patients, doctors and society at large, including offering unconditional access to its trial protocols, all additional documents such as data monitoring committees charters (which often contain information on stopping rules), investigators’ brochure, contracts between sponsor and investigators, raw anonymised patient data and case report forms. 15