
Abstract

Personalised cancer care has great appeal for many reasons. Because it presumes that one tailors specific treatments for the right cancers, personalised care implies that doctors will be providing more effective treatments with less toxic effects. Because it predicates that one identifies the various subtypes of cancer, personalised care suggests that scientists will have resolved the complexity of cancer.
A fundamental premise of personalised care is that cancer subtypes can be discerned using unique genetic and molecular biomarkers or signatures. Considering that epigenetics and the microenvironment also play an essential role in the final manifestation of the malignant process, we propose that a more comprehensive or unified theory of cancer is needed to attain personalised cancer care, in which one targets not only the genetic components but also the entire cellular make-up of individual cancer subtypes.
Personalised care: a brief history of cancer cures
In many respects, personalised care is already being practised to treat several curable cancers, such as testicular non-seminomatous germ cell tumour, acute promyelocytic leukaemia and diffuse large B-cell lymphoma. Because one can clinically distinguish these malignant subtypes, it is feasible to individualise care and assign specific treatments.
Friedman and Moore 1 classified four different subtypes of testicular germ cell tumours. Since 1980, chemotherapy has revolutionised the treatment of advanced germ cell tumours; 2 however, surgery is still indispensable for the treatment of teratomas, and both chemotherapy and surgery are required to cure mixed germ cell tumours containing teratomas.
The French–American–British classification system categorised six subtypes of acute myelocytic leukaemia. 3 Since 1991, all trans-retinoic acid and arsenic trioxide have transformed the treatment of acute promyelocytic leukaemia.4,5 Although more than 80% of patients with acute promyelocytic leukaemia experience a complete remission with all trans-retinoic acid or arsenic trioxide, fewer than 10% will do if all acute myelocytic leukaemias have been treated as one disease with the same treatment.
The Working formulation first divided non-Hodgkin lymphoma into four subtypes. 6 Subsequently, the Revised European–American Lymphoma Classification 7 used morphology, immunophenotype, genetic characteristics and clinical features to distinguish 14 distinct non-Hodgkin B-cell lymphomas. Today, chemotherapy cures about 60% of patients with advanced diffuse large B-cell lymphoma. 8
In 2001, the Human Genome Project was launched.9,10 An unspoken goal of this project was personalised medicine: When one could treat a certain cancer subtype on the basis of cancer genetics, personalised care would replace empiric care. No longer would doctors prescribe treatments by trial-and-errors or hit-and-misses. It is time to forsake the concept of a ‘one-treatment-fits-all’ approach.
Unlike testicular cancer, acute leukaemia or aggressive lymphoma, not many malignant tumours have been cured in a substantive way since 2001. We need a strategy to move above and beyond the conventional thinking of cancer genetics in an effort to develop curative targeted therapies and personalised care.
Cancer genetics
When one considers key genetic targets in cancer, a pattern emerges. In general, oncology recapitulates ontogeny. Interestingly, an oncogene is actually a defective proto-oncogene. And a malignant cell has all the characteristics of an aberrant stem cell.11–14 Hence, many key genetic targets in cancer involve stemness pathways, including pluripotency, differentiation, self-renewal and asymmetric division. Importantly, when it concerns stemness pathways, intercellular interactions and the microenvironment become integral factors in a complete equation of cancer. This interconnectedness constitutes a vital tenet of the unified theory of cancer.
For example, tyrosine kinases (e.g. Epidermal growth factor, Fibroblast growth factor and Vascular endothelial growth factor) are better known as growth factors in adult organisms, but they also play critical roles as morphogens during development. 15 The tyrosine kinase families are evolutionarily conserved from fruit fly to man and serve as master regulators of position-dependent cell fate during embryogenesis. Hence, genetic targets involved in morphogenesis are also engaged in morphostasis. The importance of morphogenetic pathways, such as RAS-RAF-MAP kinase, Phosphoinositide 3-kinase and AKT, for the maintenance of epithelial homeostasis is underscored by the fact that mutations disrupting them contribute to carcinogenesis.
Genetic targets and targeted therapies.
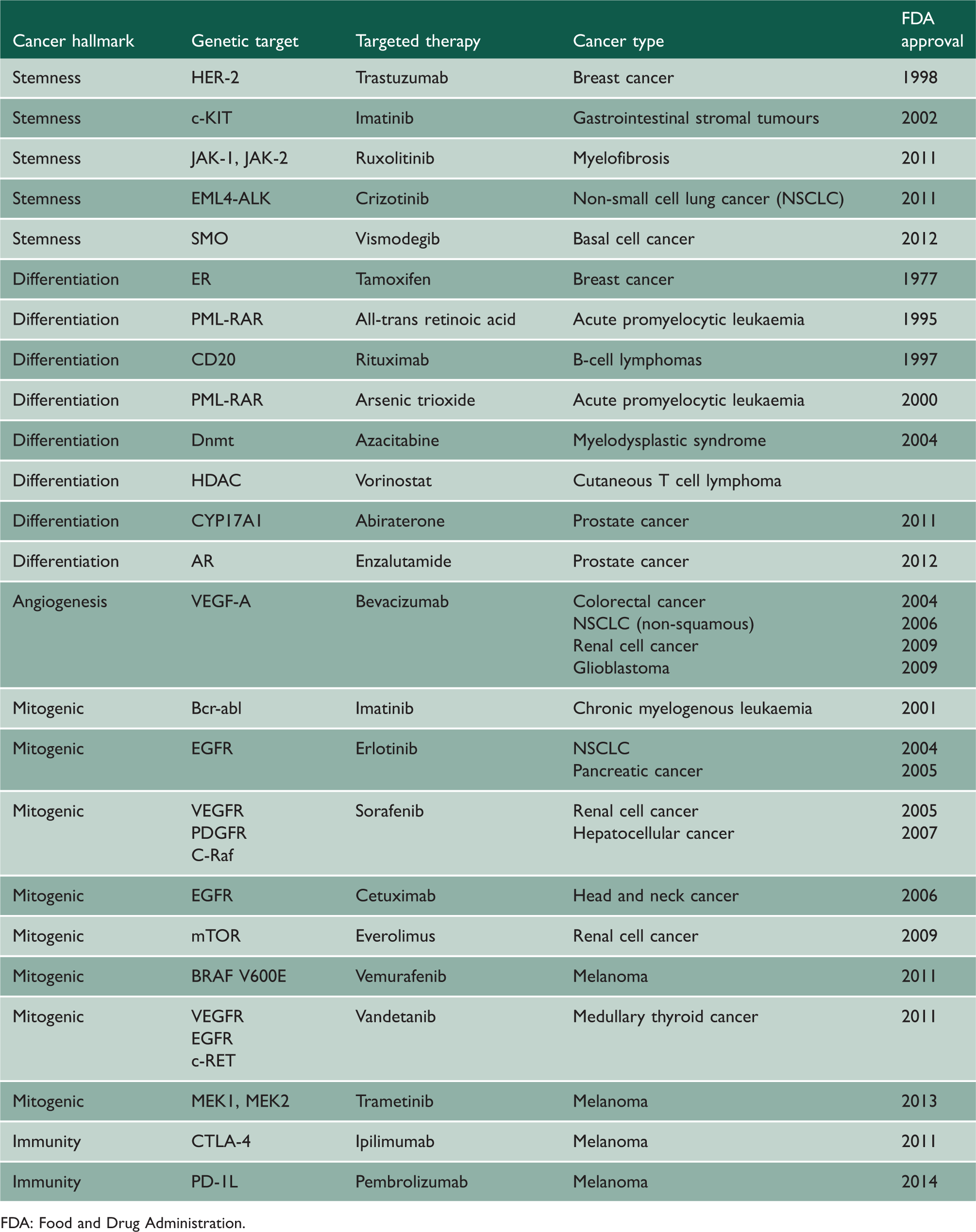
FDA: Food and Drug Administration.
Targeted therapy
The idea that cancer subtypes are better defined by their cells of origin than by their specific genetic defects is supported by clinical evidence showing a paucity of reliable biomarkers and curative therapies when the targets are based on genetic defects rather than cellular origins. Perhaps, one needs to target a whole network of genetic signatures rather than a few selected genetic defects. But when one deals with a whole network of genetic signatures, a distinct cellular entity with a unique cellular origin becomes manifested.
Considering that p53 is the most commonly mutated cancer gene in humans, one would expect p53-targeted therapy to be a boon to cancer therapy. However, the results of a phase III study did not support the prognostic value of p53 or the benefit of adjuvant methotrexate, vinblastine, doxorubicin, cisplatin chemotherapy in patients with p53-positive bladder cancer. 22 A pilot study using ONYX-015 for the treatment of oral dysplasia showed that 37% of lesions underwent histologic regression, but the responses were short-lived. 23 In addition, RAS pathway mutations occur in >90% of pancreatic cancers. However, cetuximab and gemcitabine did not improve the survival duration of patients with metastatic pancreatic cancer compared with gemcitabine alone. 24 Thus, a disconnect seems to exist between targeted therapy and its most heralded targets.
Perhaps an alternative unified theory of cancer is needed to account for the many contrary findings. For example, patients with human epidermal growth factor receptor 2-positive tumours benefited from adjuvant trastuzumab, but so did those with human epidermal growth factor receptor 2-negative tumours. 25 Such finding contradicts the very essence of targeted therapy or personalised care. Indeed, there is evidence linking human epidermal growth factor receptor 2 to cellular stemness, suggesting that it plays a central role in the intrinsic regulatory pathways of breast cancer stem cells and their interaction with the tumour microenvironment. 26 Similarly, olaparib (a poly ADP-ribose polymerase inhibitor) resulted in a 41% objective response rate in patients with BRCA1 or BRCA2 mutations but also a 24% rate in those without the mutations. 27 Thus, it seems that targeted therapy is sometimes effective regardless of whether its putative target is present or not.
One would expect tumours with plentiful mutations to be generally more lethal than those with few mutations and tumours with certain mutations to be potentially more threatening than those without them. Paradoxically, more aggressive tumour subtypes may contain fewer mutations than their less aggressive counterparts.28–30 Furthermore, the prognosis of patients with colon cancer and ‘microsatellite instability’ tumours was found to be better than that of patients with microsatellite-stable tumours. 31 And patients with advanced Epidermal growth factor receptor-overexpressing non-small cell lung cancer had longer overall survival durations than those with normal Epidermal growth factor receptor-functioning tumours.32,33 In the case of ependymonas, there are actually no gene mutations that could account for the presence or cause of a malignancy or that could be used as a prognostic biomarker or targeted therapy for personalised cancer care. 34
Promises and challenges
An inconvenient truth about targeted therapy is that the results have not been commensurate with expectations. Although the promises of targeted therapy are self-evident, its challenges are not as obvious. Perhaps, any tangible clinical benefits derived from personalised cancer care have been modest or marginal, because the fundamental concepts of targeted therapy are not aligned with a unified theory of cancer. To fulfill the promises and meet the challenges of targeted therapy and personalised cancer care, one needs to return to the basics of cancer biology. As a starter, one needs to unravel the enigma of driver mutations, intratumoural heterogeneity and cancer subtypes.
Driver mutations
When it is necessary to pick among a plethora of genetic targets, a reliable method is needed to prioritise which ones have the best chance of benefiting patients in a meaningful way, i.e. in a way that is clinically significant, not just statistically significant. Nowadays, one realises that it is not feasible to interrogate all genetic targets, many of which are incidental rather than causal, and for which treatments may prove to be futile or even harmful. It is imperative that one devises a method to distinguish the so-called driver from passenger mutations, and discerns those genetic targets that are true makers rather than mere markers of the malignant processes.
The scientific method (i.e. pertinent observation, useful hypothesis, informative experiments and empirical validations) 35 may enable one to determine whether a genetic target is the cause or an effect of cancer. Paradoxically, the current modus operandi of targeted therapy seems to be intrinsically incongruous with the fundamental principles of the scientific method. First of all, all pertinent observations indicate that cancer is a complex disease rather than a simple entity. In addition, many useful theories suggest that cancer is a multifaceted problem (e.g. genetics, epigenetics and microenvironment) in need of an integrated solution. Furthermore, informative experiments repeatedly demonstrate that several important cancer hallmarks, including metastasis, heterogeneity and dormancy, do not actually depend on any specific genetic mutations.36–41 In light of this irony, targeted therapy seems to be an oversimplification and driver mutations may be a myth.
Another method to determine whether a genetic target is the cause or an effect of cancer is to perform multivariant analysis 42 of composite genetic targets. Although targeted therapy is the product of a reductionist approach based on the traditional method of dissecting the whole and examining the parts in the laboratory, one cannot help but observe that cancer is inherently heterogeneous, interactive and dynamic in the clinic. Whereas a particular genetic target may be the ‘driver’ in isolation, it could be but one of the many ‘passengers’ in toto. By considering the whole system and its web of intricate networks, one could apply multivariant analysis to discover the real-driver mutations and design breakthrough rather than incremental targeted therapies for the benefit of personalised cancer care.
Intratumoural heterogeneity
Elucidating intratumoural heterogeneity is tantamount to overcome the challenges of targeted therapy and personalised cancer care. If intratumoural heterogeneity arises from the action of defective genes, then one targets and fixes them. On the other hand, if intratumoural heterogeneity is determined by unique cellular (e.g. stem-like) entities in which these genetic changes occur, then one needs to treat both the aberrant pluripotent progenitor cells and the diverse differentiated cancer cells.
For example, a classic targeted therapy that many people forget is androgen deprivation for the treatment of prostate cancer. 43 Almost all patients, including those with advanced metastatic disease, respond to the treatment often for years, sometimes for decades. However, androgen deprivation therapy is not a curative treatment, even though it is effective. To improve on this prototype therapy that targets differentiated prostate cancer cells, one needs to integrate it with treatments that affect other tumour compartments (e.g. prostate cancer stem cells) and the ubiquitous tumour microenvironment (e.g. bone stromal cells).
Testicular cancer provides another example of intratumoural heterogeneity in solid tumours. Because of clonal origin, it is well known that the different histologic components of germ cell tumour have a similar, if not the same, genetic profile.44–46 Hence, both chemo-sensitive embryonal carcinoma and chemo-resistant teratoma in a mixed germ cell tumour have the same wild-type p53 or a defective B-cell lymphoma 2.47,48 When a genetic mutation is observed in both the cancer stem cell and the differentiated cancer cell within the same tumour, and yet the disparate cells have distinct biological and clinical characteristics, it seems fitting that targeting the disparate cells rather than the mutation itself would be more efficacious.49,50 Indeed, by treating the distinct cell types rather than the specific genetic mutations, one has rendered testicular cancer the most curable solid tumours known to date.
Recently, several investigators showed that intratumour heterogeneity can compromise the value of biomarkers, as determined by using single tumour biopsy samples.51–53 The results suggested that genetic approach may be inadequate for solving the problem of tumour heterogeneity and for reaching our goal of personalised cancer care. Kreso et al. 54 also demonstrated that individual cancer cells within a uniform genetic lineage were functionally heterogeneous with respect to their growth dynamics, persistence through serial transplantation and response to therapy. Thus, non-genetic processes (altered epigenetic states and microenvironmental influences) also generate heterogeneity.
Therefore, personalised cancer care may be more effective when aberrant cells are targeted rather than defective genes. This approach implies that one should target the microenvironment of aberrant cells and the aberrant cells themselves, as they contain tens, hundreds or even thousands of relevant and irrelevant defective genes.55,56 When one targets cancer cells at their core, namely their cell of origin, both cancer stem cells and differentiated cancer cells are being accounted for. When one treats the intracellular, intercellular and microenvironmental factors all at once, the presence of incidental or redundant genetic defects becomes less of a concern.
Cancer subtypes
Solving intertumoural heterogeneity is another prerequisite to fulfilling the promises of targeted therapy and personalised cancer care. One way to accomplish this task is to identify cancer subtypes. By identifying subtypes, one renders a complex disease into simple ones, which can then be targeted. A popular strategy for identifying cancer subtypes is to use biomarkers. An alternative is to decipher their cells of origin. Recent evidence suggests that the key to unlocking the secrets of intertumoural heterogeneity may be found in the cellular profiles rather than the molecular genetics of cancer.
The Cancer Genome Atlas Network 57 used six different technology platforms (genomic deoxyribonucleic acid copy number arrays, deoxyribonucleic acid methylation, exome sequencing, messenger ribonucleic acid arrays, micro-ribonucleic acid sequencing and reverse-phase protein arrays) to analyse the genes of 825 breast cancer patients and confirmed the existence of four major subtypes of breast cancer: luminal A, luminal B, basal-like (triple-negative) and human epidermal growth factor receptor 2 enriched. Clinically observable plasticity and heterogeneity occur within but not across these major biological subtypes of breast cancer, suggesting that they have distinct cellular origins. Although there is enrichment of specific mutations within the tumour subtypes (i.e. phosphatidylinositol-4,5-bisphosphate 3-kinase [49%] and Mitogen-Activated Protein Kinase Kinase Kinase 1 [14%] in luminal A and TP53 [84%] in basal-like), it is uncommon for individual tumours to harbour the same genetic mutations even within the same subtype. 57 Interestingly, only about 50% of clinically human epidermal growth factor receptor 2 positive tumours fall into the human epidermal growth factor receptor 2 enriched-messenger ribonucleic acid subtype/human epidermal growth factor receptor 2-protein group, while the rest of the clinically human epidermal growth factor receptor 2 positive tumours is predominantly observed in the luminal-messenger ribonucleic acid subtypes. 57
Medulloblastoma can be parsed into unique subtypes with associated risk stratifications: Wnt pathway-active, excellent prognosis; Hedgehog pathway-active, good prognosis; and c-MYC amplification, poor prognosis. 58 Since all subtypes (i.e. different diseases) showed elevated MYC expression (i.e. the same target), it seems that the cell of origin, rather than a particular molecular defect, is paramount in the manifestation of a malignant phenotype. Indeed, Gibson et al. 59 showed that the subtypes of medulloblastoma have distinct developmental origins: the Wnt subtype arises from the lower rhombic lip and embryonic dorsal brainstem, outside the cerebellum, whereas the Sonic Hedgehog subtype originates from the upper rhombic lip and developing cerebellum.
Brannon et al. 60 surmised that the heterogeneity of clear cell renal cell carcinoma could be traced to multistep carcinogenesis, i.e. acquisition of the Von Hippel-Lindau mutation, Hypoxia-inducible factor stabilisation and secondary mutations. However, the identification of two unique renal cell carcinoma subtypes with distinct survival patterns suggests that ccB arose from earlier progenitor cells (overexpression of genes associated with Epithelial -mesenchymal transition, Transforming growth factor-β, Wnt and wound healing), whereas ccA originated from later progenitor cells (association with genes involved in angiogenesis and fatty acid metabolism). 60 Importantly, the Von Hippel-Lindau mutation itself did not differentiate between the two subtypes.
There are two distinct subtypes of posterior fossa ependymona: posterior fossa group B occurs in older children and adults and carries a very good prognosis, whereas posterior fossa group A is mainly seen in infants and is associated with poor prognosis despite intensive therapies. Different radial glial cells from different regions of the nervous system have been shown to be the cells of origin for the different subtypes of ependymonas. 61 Importantly, whole genome and whole exome sequencing across a broad cohort (n = 47) of posterior fossa ependymonas did not detect any significant or recurrent gene mutations that could account for the disparate subtypes of ependymona. 34
Conclusions
Although there is great promise and general acceptance in the use of cancer genomics for personalised care, we still need to reconcile the genetic with a cellular origin of cancer. We propose a unified theory of cancer to resolve the dilemma of targeted therapy for personalised cancer care. We advocate an integrated approach that considers both the genetic and a cellular origin of cancer to fulfill the promises and meet the challenges of targeted therapy and personalised cancer care.