
Abstract
Introduction:
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity worldwide. In addition, TBI may cause paroxysmal sympathetic hyperactivity (PSH), which is associated with poor clinical outcomes. This study aimed to evaluate the safety and effectiveness of clonidine in patients with TBI and suspected PSH.
Methods:
A retrospective cohort study for critically ill patients with TBI with suspected PSH admitted to intensive care units (ICUs) from 1 May 2016 to 31 January 2020 at a tertiary academic medical center. Eligible patients were categorized based on clonidine use during their ICU stay (Clonidine group vs. Control group). The primary outcome was the improvement in functional outcomes during ICU stay, defined by a delta Glasgow Coma Score (GCS). Secondary outcomes included ICU and hospital length of stay, heart rate variation, and 90-day mortality.
Results:
A total of 2915 patients were screened, of which 169 were included. Based on multiple regression analysis, patients who received clonidine showed better improvement in functional outcomes by a higher mean delta GCS than patients who did not (Beta Coeff. 0.41; CI: 0.07 – 0.74; P = 0.02). In addition, the patient's GCS upon ICU discharge and IV opioids requirement on day three were higher in the clonidine group than control (beta coefficient (95% CI): 0.18 (0.03, 0.32); p = 0.02 and beta coefficient (95% CI): 1.38 (0.24, 2.52); p = 0.02, respectively). No statistical differences were observed in any of the other secondary outcomes after adjusting for confounders.
Conclusion:
This study found that patients who received clonidine had better functional outcomes during their ICU stay, as shown by their delta GCS than those who did not. Other outcomes were similar between the groups. More data are needed to explore the role of clonidine in patients with TBI with suspected PSH.
Introduction
Traumatic brain injury (TBI) remains a leading cause of mortality and morbidity worldwide.1,2 Particularly, trauma secondary to a motor vehicle accident, which may result in TBI or major trauma, is the second leading cause of death in Saudi Arabia after ischemic heart disease. 3 TBI can be classified based on severity as mild, moderate, and severe based on multiple factors, including the loss or decreased level of consciousness and post-traumatic amnesia.1–3 Neurological deficits following TBI result from either primary or secondary injury.1,2,4 TBI may cause paroxysmal sympathetic hyperactivity (PSH) and catecholamine surge, both of which are linked to poor outcomes.4,5 PSH is a clinical syndrome predominantly described after TBI, which is associated with transient increases in sympathetic activity. 4 This condition is characterized by hyperthermia, tachycardia, tachypnea, and even motor features such as extensor/flexion posturing.4,5 The pathophysiology of PSH is not fully understood, but recent advances in clinical and diagnostic features have led to a broad clinical consensus, providing a foundation for systematic research on this disorder. It is a disease of exclusion and is defined as dysregulation in the autonomic function system and manifests as tachycardia and tachypnea.1,2,4,5 The incidence of PSH ranges between 8% and 33% after TBI and 6% after other brain injury causes. One study showed that the incidence of meeting criteria for PSH decreased from 24% at day 7 postinjury to 8% at 2 weeks, indicating that the timing of assessment has an impact on the difference in incidence. 4
Early identification and cessation of PSH or catecholamine surges are crucial to stop further brain injuries. 2 Multiple medications have been studied to reduce or inhibit PSH, including opioids, β-adrenergic blockers (BBs), benzodiazepines, neuromodulators, anesthetics, and alpha-2 agonists.1,2,6 Opioids are currently the mainstay therapy for PSH to aid in the resolution of hypertension, tachycardia, and allodynia.1,2,6 BBs have been shown to reduce mortality after TBI and may also be effective in PSH management.1,2,6,7 Benzodiazepines are mainly used to manage agitation, posturing, tachycardia, and hypertension.1,2,6 Neuromodulators (e.g. gabapentin and baclofen) effectively control hyperpyrexia and reduce spasticity and pain.1,2 Alpha-2 agonists reduce sympathetic outflow.1,2,6
Clonidine works as an alpha-2 agonist in the brainstem. 8 This leads to a reduction of the sympathetic outflow. 8 The most relevant safety concern with clonidine use is symptomatic sinus bradycardia and hypotension. 9 The reduction of autonomic dysregulation and sympathetic outflow were associated with mortality reduction among patients with TBI when BBs were used.7,9–11 Clonidine has a similar effect and reduces both sympathetic outflow and agitation. This reduction is desirable to help mitigate sympathetic surges in patients with TBI.1,2,8 However, limited data exist to support the use of clonidine following TBI, and guidelines make no specific recommendation regarding clonidine use.2,12–14 Therefore, the aim of this study was to evaluate the safety and effectiveness of enteral clonidine use among adult patients with TBI with agitated behavior.
Methods
This was a retrospective cohort study at the King Abdulaziz Medical City, National Guard Health Affairs (NGHA), a tertiary teaching hospital in Riyadh, Saudi Arabia. Data were obtained from electronic medical records (BESTCare) for all patients admitted with TBI for at least 24 hours from January 5, 2016 to January 1, 2020 with suspected PSH. Institutional review board approval for this study was granted by the King Abdullah International Medical Research Center (KAIMRC) (study number NRC21R/052/02) and waived the requirement for the informed consent form based on the study design. All adult patients (≥18 years) with a documented diagnosis of TBI (both penetrating and blunt) and required an intensive care unit (ICU) admission were assessed for eligibility. We excluded patients who had documented TBI without suspected PSH requiring ICU admission and TBI patients who used clonidine to treat hypertension during their ICU stay. Eligible patients were categorized based on Clonidine use during their ICU stay (Clonidine vs. Control). All patients were followed until they were discharged or died during the in-hospital stay.
Demographic data collected at baseline included age, sex, body mass index, serum creatinine (µmol/L), creatinine clearance based on the Cockcroft-Gault equation, history of hypertension, diabetes mellitus, and TBI secondary complications including seizure, brain edema, and herniation. Patient data obtained upon admission included Glasgow coma score (GCS), temperature, hypothermia (core temperature < 36.5 °C), heart rate (HR), TBI-IMPACT score, pupil size, and reactivity, Richmond agitation-sedation scale (RASS), Charlson comorbidity index, and type of head injury and type of bleeding. It is common practice at our institution to prescribe clonidine for the management of catecholamine surges in patients with TBI; thus, all patients who were prescribed clonidine had been included unless clonidine was used to treat hypertension. Follow-up assessments upon ICU discharge included GCS, heart rate, temperature, and any documented infection during ICU admission. Pertinent medication information included sedatives, antiepileptic use, and intravenous opioid dose in morphine equivalence on day 3. The co-administration of beta-blockers and quetiapine during ICU admission was also documented. Patient functional status at discharge was classified into those who were discharged without assisted devices or those who were discharged in wheelchairs or walkers. The primary endpoint was the improvement in functional status defined by the delta GCS (GCS upon ICU discharge and admission). Secondary endpoints included delta heart rate (HR) (HR upon admission and ICU discharge), RASS score upon ICU discharge, temperature upon ICU discharge, intravenous opioid dose in morphine equivalence on day 3, hospital length of stay (LOS), ICU LOS, and 90-day mortality.
The baseline characteristics of the participants are presented as mean ± standard deviation (SD) or median (interquartile range (IQR)) for continuous variables and numbers (proportions) for categorical variables, as appropriate. Continuous data were analyzed using Student's t-test or the Mann–Whitney U test. Chi-square and Fisher's exact tests were used to compare categorical data, as appropriate, and statistical significance was defined as p ≤ 0.05. Normality assumptions were assessed for all numerical variables using statistical tests (i.e. Shapiro–Wilk test) and graphical representations (i.e. histograms and Q-Q plots). Model fit was assessed using the Hosmer-Lemeshow goodness-of-fit test. For nominal outcome, multivariate logistic regression analysis was used to calculate odds ratios (OR) and p-values based on patient age, GCS score upon admission, and beta-blocker use. A generalized linear model was used to calculate the beta coefficient (estimates) and p-value based on patient age, GCS upon admission, and beta-blocker use for numerical outcomes. The OR, or estimates with 95% confidence intervals (CI) were reported as appropriate. No imputation was performed for missing data, as the cohort of patients in our study was not derived from random selection. Statistical significance was set at P < 0.05, and Statistical Analysis System (SAS) version 9.4 for all statistical analyses.
Results
Patient characteristics
A total of 2915 patients were screened, of which 169 were included. One hundred and four patients received clonidine during the ICU stay, as shown in Figure. 1. The median time for clonidine initiation was 7.0 (4.0, 9.0) days from ICU admission, with a median dose of 0.1 mg/dose (IQR: 0.1–0.2) given three times daily. Patients in the clonidine group continued on clonidine for a median of 18 days (IQR: 8.5–48), while 41.3% of them needed clonidine dose up-titration. Forty-five patients were discharged from the hospital with clonidine or other adjunctive agents, such as quetiapine, propranolol, or gabapentin. Table 1 shows the baseline characteristics of the clonidine and control groups. Patients who received clonidine were younger, had major trauma combined with TBI, lower GCS upon admission, and higher co-administration of dexmedetomidine, beta-blockers, and quetiapine as adjunctive therapy during ICU stay. Moreover, patients who did not receive clonidine (control) had higher phenytoin use as anti-seizure prophylaxis and lighter sedation within the first 24 h of ICU admission. The other variables were not significantly different between the two groups.

Flowchart of patient inclusion in the study.
Baseline characteristics.
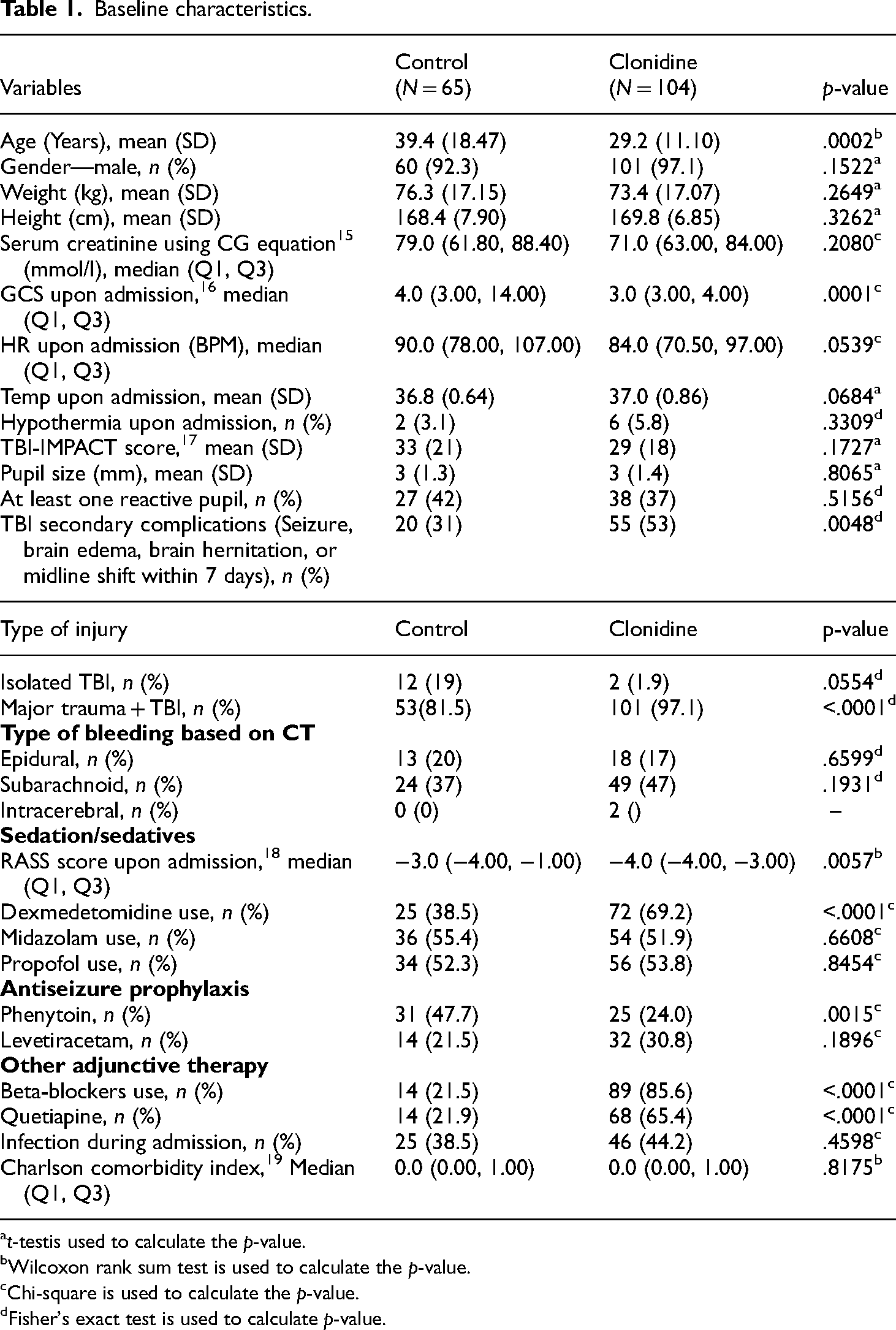
t-testis used to calculate the p-value.
Wilcoxon rank sum test is used to calculate the p-value.
Chi-square is used to calculate the p-value.
Fisher's exact test is used to calculate p-value.
Outcomes
In the crude analysis, patients who received clonidine during ICU stay had a higher mean delta GCS score than patients who did not (6.2 vs. 3.1; p < 0.01). Data remained statistically significant after adjusting for confounding variables by implementing the negative binomial regression analysis (beta coefficient (95% CI): 0.41 (0.07, 0.74), p = 0.02). In addition, the patient's GCS upon ICU discharge and IV opioids requirement on day three were higher in the clonidine group than control after adjustment of pre-defined selected variables (beta coefficient (95% CI): 0.18 (0.03, 0.32); p = 0.02 and beta coefficient (95% CI): 1.38 (0.24, 2.52); p = 0.02, respectively) (Table 2).
Regression analysis for the outcomes.
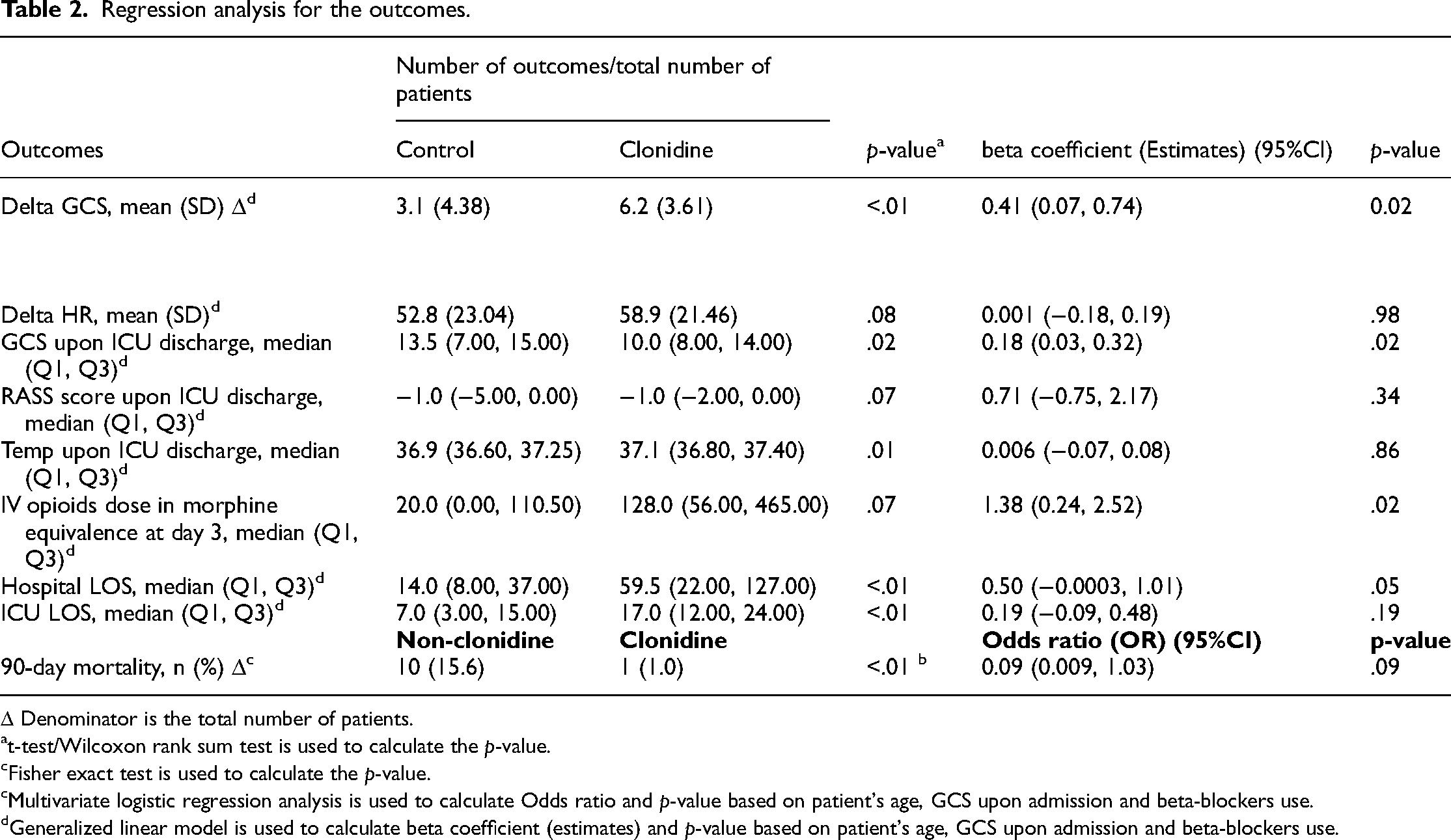
Δ Denominator is the total number of patients.
t-test/Wilcoxon rank sum test is used to calculate the p-value.
Fisher exact test is used to calculate the p-value.
Multivariate logistic regression analysis is used to calculate Odds ratio and p-value based on patient's age, GCS upon admission and beta-blockers use.
Generalized linear model is used to calculate beta coefficient (estimates) and p-value based on patient's age, GCS upon admission and beta-blockers use.
The delta heart rate was lower in the control group but did not reach statistical significance (52.8 vs. 58.9 beats per minute; p-value = 0.08). In addition, patients who did not receive clonidine had a lower median temperature upon ICU discharge or mortality, whichever occured first at crude analysis (36.9 vs. 37.1; p = 0.01). However, after adjusting for possible confounders in the regression analysis, the results were not statistically significant (Table 2).
Patients who received clonidine had a longer ICU and hospital LOS in the crude analysis; nevertheless, no statistically significant in the regression analysis was observed (beta coefficient (95% CI): 0.19 (−0.09, 0.48), p = 0.19 and (95% CI): 0.50 (−0.0003, 1.01), p = 0.05, respectively) as shown in Table 2. Other outcomes such as 90-day mortality and functional status upon discharge were not statistically significant between the two groups after implementing the regression model (OR 0.09; 95% CI 0.009, 1.03, p = 0.09 and OR 4.07; 95% CI 0.13, 124.8, p = 0.42, respectively) (Table 2). For functional status at discharge, forty-nine patients were discharged without wheelchairs/walkers, of whom 26 (53%) received clonidine (p = 0.19). Two patients who received clonidine were discharged with a wheelchair or walker.

Comparison between clonidine group vs. control group in mortality, ICU, and hospital stay and GCS improvement.
Discussion
In this retrospective cohort study, we observed better functional outcomes as evidenced by a higher delta GCS in the clonidine group than in the control group during their ICU stay. However, no difference in mortality or ICU or hospital LOS was observed in our study after adjusting for major variables.
Clonidine works as an alpha-2 agonist in the brainstem, where it attenuates the catecholamine surge following brain injury by enhancing brainstem sympathetic suppression. It mitigates peripheral resistance, renal vascular resistance, heart rate, and blood pressure; consequently, the excessive sympathetic outflow decreases. 20 Moreover, two of the main manifestations of PSH are tachycardia and hyperthermia. Our study results showed higher delta heart rate and temperature in the clonidine group; however, this did not reach statistical significance. Similarly, Megahed et al. found no statistically significant difference in the mean temperature between the two groups; however, the heart rate was significantly reduced in the interventional group. 13
This result is in contrast to a study conducted by Megahed et al., who found no significant difference in GCS score after administering clonidine in addition to propranolol in PSH after severe TBI. 13 In which longer ICU and hospital LOS were observed in the crude analysis but were not statistically significant in the regression analysis. Likewise, Barmparas et al. found that patients who received beta-blockers had a longer LOS than those who did not. 10 Despite the increased co-administration of beta-blockers in the clonidine group compared with the non-clonidine group. Our study demonstrated similar clinical outcomes in hospital and ICU LOS.
Moreover, this study found no mortality benefit with clonidine administration, which is consistent with previously published data. 13 It is unclear why previous studies demonstrated the potential mortality benefit of using beta-blockers while clonidine did not. A possible explanation could be the timing of clonidine initiation, the effect of beta-blocker co-administration, and treatment duration. Additionally, titration of clonidine is challenging because of the risk of inducing hypotension in the initiation phase and the imposed risk of reflexive hypertension in the discontinuation phase.8,20 It is worth mentioning that 70% of patients in the clonidine group in our study had received dexmedetomidine compared to 38% in the non-clonidine group, which can impact the results when considering dexmedetomidine withdrawal.21,22 These outcomes might cause clinicians to have clonidine reserved for refractory PSH cases that may not respond properly to beta-blockers alone.8,10–25 Patients in the non-clonidine groups received less IV opioids compared to the clonidine group, which can be related to the baseline risk. This can be further explored to assess the analgesic impact of clonidine in patients with TBI. Notably, we used mean and SD to report our delta GCS for the whole cohort, which was deemed to have a normal distribution. Thus, calculating delta GCS by using the difference in median GCS admission and discharge would result in erroneous numbers.
This study had several limitations. First, the retrospective method used in this study might have contributed to selection bias or confounders affecting the results despite the use of a regression model. Second, the bias size may be too small to detect statistical differences, which decreases the power of our study. Furthermore, our study was conducted in a single center, contributing to the small number of included patients and limited its generalizability. Moreover, the nature of PSH as a rare condition with no clear diagnostic criteria following TBI has contributed to the insufficient number of patients provided by a single center. These results should be validated using a more robust study design.
Conclusion
Our study found that patients who received clonidine had better functional outcomes during their ICU stay, as shown by their delta GCS than those who did not receive clonidine. Furthermore, it was observed that upon leaving the ICU, patients in the clonidine group had higher Glasgow Coma Scale (GCS) scores and required more intravenous opioids on the third day compared to those in the control group (beta coefficient (95% CI): 0.18 (0.03, 0.32); p = 0.02 and beta coefficient (95% CI): 1.38 (0.24, 2.52); p = 0.02, respectively). However, other outcomes, such as in-hospital mortality, improvement in PSH symptoms, and ICU and hospital LOS, were not significantly different between the two groups. Further investigations are needed to identify the role of clonidine in PSH following TBI.
Footnotes
Acknowledgments
We want to thank all the investigators who participated in this project from the Saudi Critical Care Pharmacy Research (SCAPE) platform.
Authors’ contributions
All authors had substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work, drafting and revising the content and approving the final version.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
IRB approval for this study was granted by IRB of KAIMRC with study number NRC21R/052/02
Author biographies
Abdulrahman I Alshaya is a Consultant Critical Care Clinical Pharmacy and Associate Professor of Pharmacy Practice at the College of Pharmacy at KSAU-HS and MNGHA-CR.
Mohammed Aldhaeefi is a Clinical Assistant Professor and Critical Care Clinical Pharmacist at Howard University.
Nada Alodhaiyan is a Licensed Pharmacist.
Sara Althewaibi is a Licensed Pharmacist.
Wala Alshahrani is a Licensed Pharmacist.
Khalid Al Sulaiman is a Consultant Critical Care Clinical Pharmacy and Assistant Professor of Pharmacy Practice at the College of Pharmacy at KSAU-HS and MNGHA-CR.
Shmeylan Alharbi Consultant Critical Care Clinical Pharmacy and Professor of Pharmacy Practice at the College of Pharmacy at KSAU-HS and MNGHA-CR.
Ramesh Vishwakarma is a Senior Biostatician, European Organization for Research and Treatment of Cancer.
Tarek Aldabbagh is a Consultant Critical Care Physician, Head of Trauma Critical Care Units at MNGHA-CR.