
Abstract
Spinal muscular atrophy (SMA) is a severe, life-limiting neuromuscular condition associated with progressive disability and premature death. The condition significantly affects the quality of life of patients and their families, often resulting in psychological distress and unmet care needs. Despite growing clinical interest, qualitative evidence on the lived experiences of individuals with late-onset SMA types (II, III, IV) remains limited. This review aimed to synthesize qualitative findings on patient and family experiences in the context of complex symptomatology, unpredictable progression, burdensome caregiving, anticipatory grief, and end-of-life processes. A systematic search across EBSCO Discovery Service, Web of Science, SCOPUS, PubMed, and ProQuest identified 785 studies, of which seven met the inclusion criteria. The review followed PRISMA and SPIDER guidelines, and thematic synthesis revealed three key themes: (1) SMA as a serious condition with challenging symptomatology; (2) Severity of psychosocial impacts of SMA; (3) SMA in the context of experienced loss and premature death.
Keywords
Introduction
Spinal muscular atrophy (SMA) is a progressive, life-limiting neuromuscular condition characterized primarily by muscle weakness (Kiefer et al., 2020; Parente & Corti, 2018). SMA is one of the most serious genetic conditions, it typically presents in infancy or childhood but can also occur in adolescence or adulthood (Belter et al., 2020; Gitlin et al., 2010; Yang et al., 2016; Zerres et al., 1997). SMA is the second leading genetic cause of premature death after cystic fibrosis (Lunn & Wang, 2008; D’Amico et al., 2011) and often results in immobility and respiratory failure (Jędrzejowska, 2020). The prognosis varies depending on the type of SMA and respiratory functions (Darras, 2015; Wirth, 2000). In general, proximal and lower limb muscles are more severely affected, with symmetrical involvement. Progressive motor neuron loss leads to impaired movement, swallowing, breathing and other symptoms, depending on the type of SMA (Brandt et al., 2022; Moultrie et al., 2016; Strunk et al., 2019). Despite physical limitations, cognitive and emotional functions remain intact (Oskoui et al., 2017; von Gontard et al., 2012). SMA significantly impacts independence and quality of life, causing stress for patients and their families (Aranda-Reneo et al., 2020; de Oliveira & Araújo, 2011; Peña-Longobardo et al., 2020; Qian et al., 2015). Many patients require continuous care from medical technologies and caregivers (Farrar et al., 2017, 2018; Yang et al., 2016). The unpredictable nature of the condition and limited public awareness further complicate matters (Gregoretti et al., 2013). SMA is classified into five types (0–IV), with type 0 being prenatal and most severe, type I early-onset, and types II–IV typically late-onset (Arnold et al., 2015). Type I was long considered the most common, but types II and III now appear more frequently as children with type I often die by the age of two (Iannaccone et al., 2004). The traditional classification system is based on age of onset and death, motor milestone achievement, and ambulatory status (Chung et al., 2004). SMA type II is characterized by milder proximal muscle weakness and a later onset, typically occurring between six and 18 months of age (Ogino et al., 2002). Patients, referred to as ‘sitters’, can sit unassisted but never walk independently, though some may stand with braces (Chen, 2020). Common features include impaired swallowing, chewing, breathing and kyphoscoliosis (Arnold et al., 2015; Catteruccia et al., 2017; Kayadjanian et al., 2013), which often requires respiratory support. Feeding difficulties, hypotonia and areflexia are also prevalent. Although many patients survive into adulthood, their life expectancy is reduced (Moultrie et al., 2016) and their symptoms tend to worsen with age (Markowitz et al., 2012). End-of-life events may occur from adolescence to late adulthood (Lunn & Wang, 2008). Although the disease is mildly progressive, it often follows long stable periods (Glanzman et al., 2011). Cognitive abilities are usually unaffected (von Gontard et al., 2012), and many patients transition successfully into adulthood. However, anxiety about future decline remains a significant challenge (Jeppesen et al., 2010).
SMA type III, accounting for approximately 10% of cases, is a milder form that may present in childhood, adolescence, or adulthood (Arnold et al., 2015; Carré & Empey, 2016). Diagnosis typically occurs after age four (Dunaway et al., 2012), with symptoms such as frequent falls and fatigue appearing after age two (Kaufmann et al., 2011; Markowitz et al., 2012). Lower limbs are more affected, often leading to scoliosis, tendon shortening, and respiratory issues (Mercuri et al., 2012; Messina, 2018, 2020). Patients (often referred to as ‘walkers’) generally have normal life expectancy (Arnold et al., 2015), though some may become wheelchair-dependent in midlife (Devade et al., 2022). While they can walk unaided (Lunn & Wang, 2008), difficulties with rising, stooping, or climbing stairs are common (Iannaccone, 2007). SMA type IV, a rare adult-onset form, typically emerges in the second or third decade (Zerres & Davies, 1999), with diagnosis around age 30–35 (Chen, 2020). It presents with mild symptoms, no respiratory or feeding complications (Brandt et al., 2022), and does not affect life expectancy (Clermont et al., 1995; Piepers et al., 2008).
Until late 2016, SMA was considered incurable, with treatment limited to supportive and palliative care. However, since 2017 (and 2021 for adults), new therapies – most notably Nusinersen – have significantly altered the progression of the disease and improved survival rates (Finkel et al., 2017). These advances, alongside improvements in supportive and respiratory care, have transformed the therapeutic landscape, enhanced quality of life and reshaping the experience of the illness (Mercuri et al., 2018; Wan et al., 2019). Although a complete cure remains unavailable, survival, motor and respiratory functions can now be positively influenced (Mercuri et al., 2018; Waldrop et al., 2005, 2015).
Anticipatory grief, which is emotional distress that occurs before the actual loss, is common among families of individuals with SMA types I and II (Lindemann, 1994). It can have a profound psychological impact, often causing prolonged physical and mental fatigue (Al-Gamal, 2013; Yang et al., 2016), as well as feelings of helplessness and being overwhelmed (Rolland, 1994). This is exacerbated by the inability to influence the loved one’s condition and the fear of impending death. Following a diagnosis, parents or adult patients often experience shock, numbness, denial and growing sadness (Lawton et al., 2015; Qian et al., 2015; Yang et al., 2016). This can lead to prolonged grief and depression when confronted with the prospect of premature death and disrupted life expectations (Yang et al., 2016). Caregivers, primarily parents, undergo intensive training to provide daily home care (Di Pede et al., 2018). Integrating pediatric palliative care is essential, particularly for severe SMA types (Mitchell, 2006; Mitchell et al., 2017), with a focus on home care and minimizing hospitalizations (Wang et al., 2007). In Higgs et al. (2016), parents expressed concern about their child’s suffering and viewed death as a form of relief. They also valued decision-making around end-of-life care. Communication styles influence grief outcomes: open sharing fosters closeness and comfort (Bergstraesser et al., 2015), whereas avoidance can lead to more complicated grief (Stroebe et al., 2013; Stroebe et al., 2007). Nevertheless, the end-of-life experiences of individuals with SMA remain under-explored. While the views of children and adolescents are often mediated by caregivers, young adults are increasingly sharing their opinions directly, making them valuable contributors to qualitative research.
Aim of the Review and Identification of the Review Question
This qualitative evidence review aimed to examine and synthesize primary research findings on the experiences, needs and expectations of individuals with late-onset SMA (types II, III and IV) and/or their families, in relation to its severity, uncertain progression, end-of-life care and bereavement. Relevant studies were identified using the SPIDER tool, with both patient and family perspectives analyzed. The review was framed by the question: What qualitative evidence exists regarding how individuals with late-onset SMA and their families experience illness progression, the end of life, bereavement, and the demands of care?
Materials and Methods
Review Design
This qualitative evidence review [hereafter referred to as ‘qualitative review’] used thematic synthesis to analyze and interpret the findings of primary research (Flemming & Noyes, 2021; Thomas & Harden, 2008). This approach is well-suited to exploring lived experiences and care-related phenomena (Evans & Pearson, 2001; Lockwood et al., 2015) and supports the development of theory and a deeper understanding of socio-cultural contexts (Purssell & McCrae, 2020; Rodrigues da Silva Noll Gonçalves et al., 2023). Meta-synthesis procedures were applied to systematically integrate qualitative evidence (Sandelowski & Barrosso, 2007; Noyes et al., 2019). This review followed PRISMA guidelines (Page et al., 2021; Shamseer et al., 2015) and utilized the SPIDER tool to define the search strategy (Methley et al., 2014) (for description, see below).
Eligibility Criteria
Eligible studies included original empirical research with primary qualitative data focused on individuals with SMA types II, III, or IV and/or their families, addressing experiences related to SMA severity, progression, end-of-life, and bereavement. Both qualitative and mixed-method studies were considered, with only the qualitative components analyzed. Inclusion was limited to full-text, peer-reviewed articles published in English between 2000 and 2024. Studies were excluded if they lacked a clear qualitative design, did not specify the SMA type (determined according to the information provided in the dedicated section of the relevant study), or focused solely on medical, psychiatric, psychological, or ethical-legal aspects of end-of-life and related topics. Abstracts, editorials, and conference papers were also excluded.
Search Strategy and Study Selection
SPIDER Framework – Database Search Strategy

The primary search terms comprised combinations of keywords for a condition [“spinal muscular atrophy” or “SMA” or “sma2” or “smaII” or “sma type 2” or “sma type II” or “sma-2” or “sma-II” or “sma3” or “smaIII” or “sma type 3” or “sma type III” or “sma- 3” or “sma-III” or “sma4” or “sma type 4” or “sma type IV” or “sma-4” or “sma-IV”] and/or the search terms “adult*” or “late-onset” or “late onset” or “SMA in adult*” or “adult SMA” or “adult form* SMA” (TI, AB) was entered into the search engine/databases, along with the search terms “palliative” or “hospice” or “end of life” or “end-of-life” or “EOL” or “dying” or “death” or “bereavement” or “grief” or “mourning” or “terminal” or “caring” or “demand*” or “exhaust*” or “family car*” or ‘distres’ or ‘cope’ or ‘coping’ (TI, AB). Terms such as ‘person(s)’, ‘patient(s)’, ‘client(s)’, ‘individual(s)’, ‘close person(s)’, ‘family member(s)’, and others were considered synonymous for the purposes of searching for relevant studies and conducting this qualitative review.
The initial search yielded 785 studies that met the criteria. After removing duplicates and studies unrelated to the aim of the review, 75 studies were retained for full-text screening. The titles and abstracts of these studies were assessed for relevance, resulting in a final sample of seven studies forming the qualitative evidence base. Figure 1 (PRISMA flow diagram) outlines the screening and selection process. PRISMA Flow Diagram of Study Selection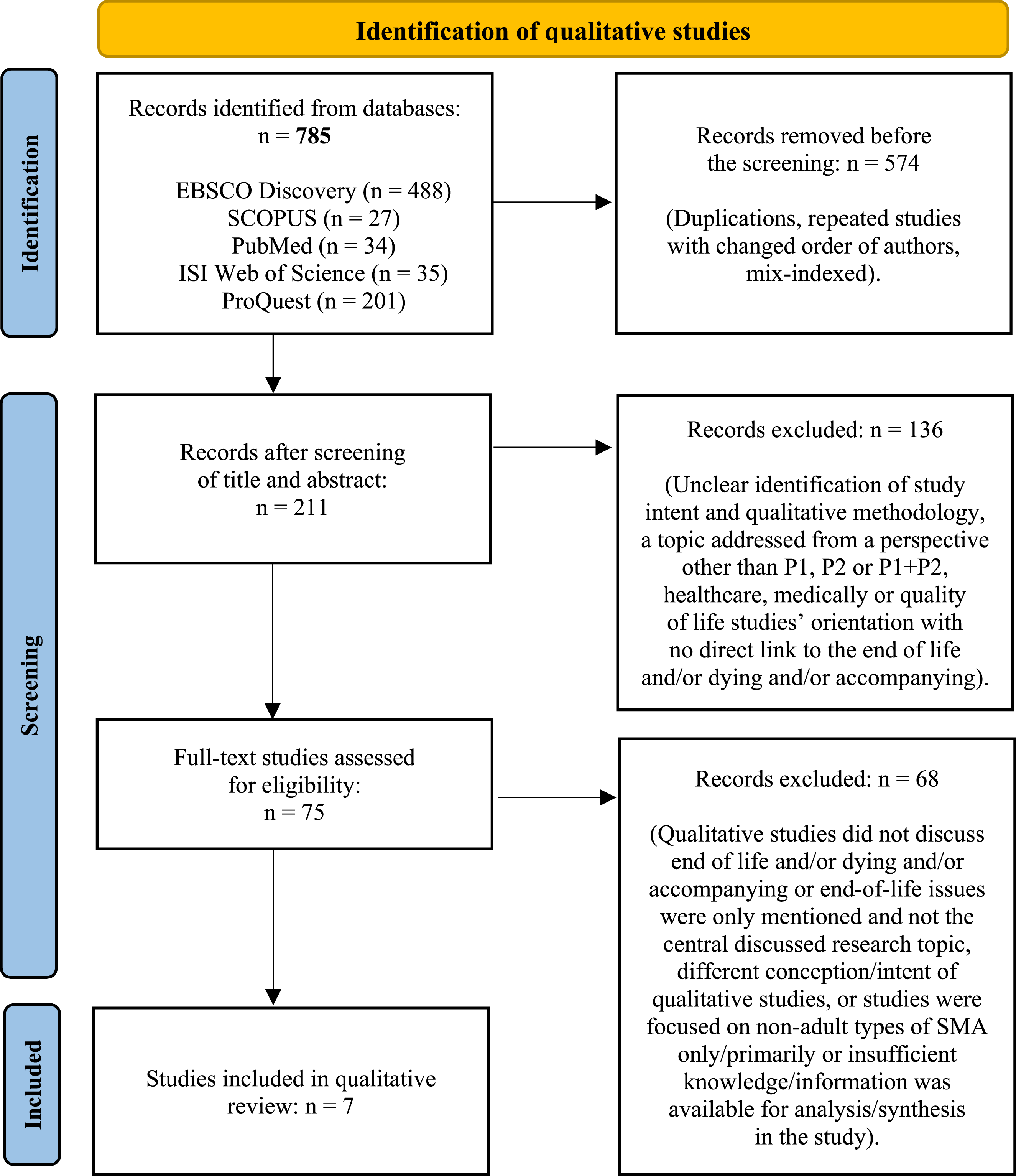
The methodological quality of included studies was independently assessed by both authors using the CASP Qualitative Studies Checklist (2018), a 10-item tool designed for evaluating qualitative research in systematic reviews. Scores ranged from 7 to 9 out of 10, indicating moderate to high methodological rigor.
Data Extraction/Analysis and Thematic Synthesis
The data extraction process began by documenting the key characteristics of the study, including the geographic context, publication year, qualitative design, sample, findings and perspective on the phenomenon. Qualitative findings from the final sample (n = 7) were systematically extracted and summarized in tables. Participant quotes were coded and annotated collaboratively by both authors to reach a consensus. The authors repeatedly read each study, reflecting on their interpretations and refining their thematic insights. A shared codebook was developed to guide the coding process and enable the organization of data into initial categories, which were then organized into subordinate and superordinate themes. Thematic synthesis was employed as the analytical framework (Braun & Clarke, 2006; Thomas & Harden, 2008), taking an inductive, bottom-up approach. First- and second-order constructs were grouped into third-order analytical themes, which were then abstracted into key interpretative themes that formed the basis of the Results section. The SPIDER tool was used to verify the alignment of the extracted findings with the focus of the review, thereby supporting the generation of transferable insights.
Results
Description of the Studies
The systematic search identified 785 records, of which seven met the eligibility criteria and were included in the qualitative review. These studies, referred to as SMA1–SMA7, were conducted across five countries: the United States (SMA1, SMA3, SMA7), Malaysia (SMA2), Taiwan (SMA4), Denmark (SMA5), and Australia (SMA6). Publication years ranged from 2008 to 2022, with the most recent study published in 2022 (SMA2), followed by studies from 2019 (SMA6), 2017 (SMA3), 2016 (SMA4), 2015 (SMA1), 2010 (SMA5), and 2008 (SMA7).
The qualitative review was conducted from the following perspectives: the person/patient with SMA themselves (SMA1, SMA2, SMA3, SMA4, SMA5, SMA6, and SMA7); family carers (reported as families, parents or partners) (SMA1, SMA2, SMA3, and SMA6); and staff/care providers (SMA1 and SMA3).
In terms of the research design, the relevant studies were designed as follows: focus groups with interviews (SMA1, SMA2, and SMA3); a phenomenological approach (SMA4); a narrative approach (SMA5); a mixed-methods design (SMA2 and SMA5); a qualitative cross-sectional study (SMA6); and a qualitative descriptive design (SMA7). The data in these studies were collected using semi-structured or in-depth interviews or qualitative interviewing (SMA1–SMA7), one of which was conducted remotely (SMA7).
Finally, analytical procedures derived from grounded theory, based on coding and categorizations (SMA1, SMA3, and SMA7), thematic analysis (SMA2, SMA6), descriptive phenomenological analysis procedures according to Colaizzi (SMA4), narrative analytical conceptions of stories (SMA5), or coding procedures with a process of constant comparison (SMA7) were used as methods of analysis.
As shown in Table 2, the following section summarizes the studies included in the qualitative review, including the CASP values. Studying, analyzing and synthesizing the qualitative findings (i.e. the evidence) resulted in the following three main themes: 1. ‘SMA as a serious condition with challenging symptomatology’. 2. ‘Severity of psychosocial impacts of SMA)’. 3. ‘SMA in the context of experienced loss and premature death’. Characteristics of Included Studies and Data Extraction ID = qualitative study identifier, CASP = Critical Appraisal Skills Programme: Qualitative Studies Checklist. aThe study included participants with SMA type I, II and III, therefore the results were only related to SMA type II and type III. bMost participants (9 out of 13) were in the age category 20–29 years. cIn this study, a ‘caregiver’ was defined as a member who provides at least 4 hours per day of care for at least one activity of daily living (ADL). dThe SMA duration median was six years (range 1–28 years). eThis was, for example, an assessment of change(s) in the inability to do activities important for more independent living. These included simple everyday tasks such as opening doors, combing hair, going to the toilet independently, being able to turn pages in a magazine, holding a book, stroking a dog with a firmer touch, holding a drink, making a phone call, and others. fAdults with SMA type II lived mostly alone and reported a good quality of life. They were mostly able to do what they wanted, had good friends and were satisfied with their lives. All had a state-funded personal assistance programme, 83 % of them continuously.

Theme 1: SMA as a Serious Condition with Challenging Symptomatology
For many individuals and families, receiving an SMA diagnosis is described as a prolonged and distressing process, often involving multiple consultations with pediatricians (in early-onset cases) or specialists (in later-onset forms) (SMA1). Although SMA is a rare disease, it is relatively common within this spectrum, and most pediatricians are likely to encounter at least one case in their career. However, limited clinical awareness may delay an accurate diagnosis and complicate the management of symptoms (SMA1). Parents of children with SMA type III reported that early signs of muscle weakness were misattributed to other conditions, such as muscular dystrophy, which further delayed appropriate care and discussions around palliative support. SMA can present with subtle symptoms, such as hand tremors or difficulty getting up from the floor, that may go unnoticed until adolescence (SMA1). Adults with SMA type II often experience pain, particularly when sitting for long periods of time, and impaired hand function, which worsens with age and exposure to the cold (SMA5). These symptoms can significantly hinder self-care and social participation. Fatigue and stress are also significant challenges in adulthood (SMA6), with the condition being described as both debilitating and transformative (SMA7). The communication and acceptance of the diagnosis were universally challenging. SMA type II was perceived as particularly burdensome due to its trajectory. SMA type III typically manifests later. SMA type I was excluded from this review due to its early onset. Caregivers reported severe sleep disturbances and even complete sleep deprivation due to the intensity of their caregiving responsibilities (SMA1). Young individuals transitioning to adult care described this process as ‘challenging and frightening’ (SMA6). One participant noted: “Sometimes they send you a carer who does not know how to use a hoist” (Wan et al., 2019, p. 4; paraphrased), highlighting gaps in continuity and competence across care systems. Navigating unfamiliar services and professionals was often perceived as impersonal and overwhelming. Despite these challenges, adults emphasized the importance of creativity and adaptability in managing symptoms (SMA7), employing physical strategies (e.g. energy conservation) and emotional coping mechanisms (e.g. social engagement and optimism).
Theme 2: Severity of Psychosocial Impacts of SMA
The emotional and social burden of SMA significantly affects families, including siblings whose needs may be overlooked (SMA1). The impact may be less pronounced in milder forms (types III and IV), whereas in early-onset types (I and II), the psychosocial strain is often substantial. Stigma and embarrassment were reported when individuals with SMA were excluded from physical activities (SMA1, SMA2, SMA6) or stereotyped and treated differently (SMA6). Limited opportunities for social engagement due to reliance on respiratory support, wheelchairs, fatigue or infection risk further contributes to isolation (SMA1, SMA2). Participants described achieving independence and autonomy as a major challenge (SMA1, SMA6), often linked to feelings of social exclusion. Participants reported difficulties with basic self-care tasks such as toileting, turning in bed, personal hygiene and feeding (SMA2), which reinforced concerns about dependency and loss of self-sufficiency. These limitations significantly affect the quality of life for individuals and their families. A gradual loss of control over daily tasks (SMA2), reduced mobility (SMA3), and challenges in maintaining head and limb positioning – or at least to maintain a (fixed) head position and to readjust this position after a flop over – were reported as key determinants of independence (SMA3). On the other hand, the demanding and frequent head positioning causes great difficulties, as does the positioning of the limbs and the adjustment of the overall body position (SMA3). Although these difficulties were substantial, adults with SMA type II rated their quality of life positively (SMA5).
The progressive nature of SMA complicates psychological adjustment (SMA1). Individuals described feelings of stress, anxiety, and depression, particularly in SMA type III (SMA2), with similar emotional burdens reported by caregivers – most notably in cases involving SMA type I. Only one caregiver sought professional psychological support, possibly due to cultural stigma surrounding mental health and emotional expression. In the Malaysian context, negative societal attitudes toward disability may further discourage help-seeking behaviour.
Individuals with SMA reported experiencing discrimination in employment, including unequal access to job opportunities and inadequate workplace support (SMA2, SMA6) or the support they needed (SMA6). Some adults engaged in part-time work (SMA4, SMA6), though the high financial burden of living with SMA remained a significant concern (SMA6). Supportive relationships with family, caregivers, and colleagues (SMA7) were valued, as was the role of social workers and psychosocial support (SMA6). Participants’ school experiences were marked by both discrimination and support. In SMA2 study, participants reported being bullied for their gait and being mislabeled as having an intellectual disability. Conversely, some described receiving meaningful support from peers and staff. Findings from SMA5 study revealed that one-third of participants were excluded from classes, which negatively affected their academic performance and transition to subsequent life stages, particularly among girls in years 7–9. It is notable that all participants attended mainstream schools and demonstrated cognitive abilities comparable to their peers.
Meaningful changes in functional ability were described by patients, caregivers, and clinicians (SMA3), with considerable individual variation – even within the same SMA subtype. While some improvement may occur, many participants expressed satisfaction with simply maintaining existing abilities. Functional decline often progresses with age, particularly in children (SMA3), yet even minimal improvements can have a profound impact. As one study noted: “the difference between not being able to move a finger and moving it half a centimeter can determine whether a person can operate a motorized wheelchair – dramatically affecting independence and quality of life” (McGraw et al., 2017, p. 4; paraphrased).
Theme 3: SMA in the Context of Experienced Loss and Premature Death
Individuals with SMA live with ongoing complications and the persistent threat of premature death. Over time, they come to understand the risks associated with their condition, learning to navigate daily challenges while striving to live in the present and appreciate life despite its limitations (SMA4). Many express a need to value everyday moments and maintain hope, even within the context of a shortened life expectancy. Patients and caregivers alike perceive life as a continuous process marked by fear of physical decline and emotional distress linked to progressive loss (SMA1). SMA poses serious threats to economic, social, and psychological independence, often creating tension between autonomy and dependence, which shapes both daily living and personal identity (SMA4). Witnessing the suffering of a child, adolescent, or adult with SMA is emotionally taxing for family members (SMA1). This burden is poignantly illustrated in the testimony of Mei, a young participant with SMA (Ho et al., 2016, p. 2700; paraphrased): “I rely on my parents for everything. When I need to use the toilet, my mother lifts me from the wheelchair to the bed, removes my trousers, carries me to the toilet, then back to the bed to dress me again, and finally returns me to the wheelchair. She does this ten times a day. It’s a huge physical burden – and now my parents are getting older.”
Loss of control was identified as a central challenge for individuals with SMA (SMA4, SMA7). Accepting the gradual loss of abilities – such as giving up social activities like dining out – was emotionally difficult (SMA1). Participants also described the psychological impact of unmet expectations and the diminishing sense of independence and autonomy (SMA4, SMA6, SMA7). One participant illustrated this tension: “Being able to decide how to live your own life and knowing what’s best for you doesn’t mean doing everything yourself – it means being the one who decides what is done and how, and knowing who does it” (Wan et al., 2019, p. 7; paraphrased).
Being dependent on others for daily care placed a significant burden on family members and carers (SMA4, SMA6). Functional decline was often accompanied by sadness and depression (SMA6), and many individuals with SMA described finding self-worth, self-esteem, and meaning in life as central to their ability to cope (SMA6). Loss of autonomy was reflected in difficulties with personal care tasks such as shaving, washing and grooming, which worsened with age (SMA5, SMA6 and SMA7). Parents of adult patients also reported challenges in providing intimate care, and most participants with SMA type II were unable to perform household tasks such as cooking, cleaning or doing the laundry.
Relationship and partnership roles were also affected. While some adults with SMA type II expressed a desire for romantic relationships, others viewed the realization of partner or parental roles positively (SMA6). However, data from the SMA5 study indicated that 73% of participants aged 30–69 were single, which is more than three times the rate in the general population (Jeppesen et al., 2010, p. 12). The significance of functional loss should not be underestimated. Individuals with SMA type II reported serious challenges related to the decline of their hand and finger function, as well as difficulties with speech, eating and breathing (SMA5). For some, denial was a coping mechanism in response to the fear of losing specific abilities (SMA6). The grieving process for lost functions was described as a form of functional loss (SMA6), which often led to social isolation and a perceived loss of normality (SMA6, SMA7).
Feelings of deep sadness, grief, and uncertainty about the future were commonly reported by both individuals with SMA and their caregivers (SMA2, SMA7). High levels of stress and anxiety, persisting from diagnosis onwards, were especially prevalent among carers. Due to the progressive nature of SMA, patients often expressed fear about the future, including concerns about losing the ability to breathe independently (SMA7). Many described a loss of normality and persistent worry about SMA progression and its impact on daily life (SMA2, SMA5). Peer support from other adults with SMA was identified as a valuable source of comfort and inspiration (SMA6). However, providing comprehensive supportive care, including palliative care, is often complicated by the central concern of functional decline, such as a reduced ability to write, operate a wheelchair, eat unaided or breathe independently. Fears about future ventilator dependence and potential cardiac complications were also noted. Uncertainty about the future was poignantly captured in the testimony of a 22-year-old participant with SMA type II: “My biggest worry now is that I won’t be able to live independently in the future… because I know my parents won’t always be here. I’m wondering if I’ll be able to find a way to live on my own when they’re gone… that’s what worries me the most” (Ch’ng et al., 2022, p. 6; paraphrased). For families who have already experienced the traumatic loss of a child to SMA, the potential death of another child represents an overwhelming and cumulative life event (SMA2). Premature death is one of the most devastating consequences of SMA, not only in type I but also in later-onset forms, where depression and thoughts of death have been reported (SMA1). A paradox emerges for patients and caregivers: planning for death while simultaneously planning for the future (SMA1). Notably, none of the participants in the reviewed studies had been referred to palliative care specialists. Adults with SMA often struggle to formulate life goals, which affects their self-concept and future outlook (SMA4). Caregivers expressed concern that their loved ones sometimes underestimated symptoms or ignored signs of deterioration (SMA6), which further complicates care planning and emotional preparedness.
Discussion
SMA is a progressive neurodegenerative condition characterized by a wide ages range at which symptoms first appear, varying symptom severity, and associated complications. It places a significant and complex burden on patients and their families. Traditionally, classification has been based on age at symptom onset and attainment of motor milestones, with five distinct types being identified (Farrar et al., 2017). However, the advent of disease-modifying therapies has significantly altered the natural history of SMA, making conventional classification frameworks inadequate. These interventions have enabled some patients to achieve developmental milestones that were previously unattainable, complicating their alignment with established diagnostic categories. Contemporary treatment can enable individuals diagnosed with SMA type II to experience a milder progression of symptoms and potentially achieve a life expectancy like that of the general population. However, the long-term sustainability of symptom reversal and the necessity for ongoing therapeutic intervention are still uncertain. Furthermore, limited research into the psychosocial and clinical needs of patients with SMA type II and those newly eligible for treatment limits our understanding of whether extended survival will reveal new age-related comorbidities in this evolving patient group (Chen, 2020).
The course of the illness is characterized by an extended and emotionally demanding diagnostic process, followed by lifelong challenges involving physical, psychological, social and financial burden (Droege et al., 2020; Mongiovi et al., 2018). Adults with SMA often experience feelings of sadness, anxiety, frustration and isolation, particularly when their physical function declines. Consequently, individuals with SMA and their caregivers require long-term multidisciplinary medical and psychosocial support to maintain mobility, improve quality of life, and promote family stability (Finkel et al., 2018; Wang et al., 2007).
Although adults account for around 35% of the global SMA population (Verhaart et al., 2017), research has largely centered on pediatric care, resulting in a lack of exploration of adult experiences (Klug et al., 2016; López-Bastida et al., 2017; Rouault et al., 2017). Therapeutic decision-making does not end with the initial diagnosis but continues throughout the course of the condition. While parents may initially be reassured by treatment progress, uncertainty can resurface when improvement stalls or disability worsens (Boursange et al., 2022). Structured guidance and anticipatory support can facilitate informed decision-making and foster trust between families and care teams.
Patients with late-onset SMA usually have normal motor development in the early stages, with physical decline emerging in adulthood. The need to continually adapt to progressive limitations often results in psychological stress, as individuals must reassess their personal goals and daily functioning (Kruitwagen-van Reenen et al., 2018). Coping strategies are crucial for maintaining participation and life satisfaction, especially for those with SMA types III and IV, who may experience difficulties with adjustment later in life. Pain is a significant and multifaceted barrier that commonly arises from spinal deformities, muscle spasms or neurogenic origins (Abresch et al., 2002).
Although patients with late-onset SMA experience fewer physical restrictions, they should not report a higher level of satisfaction regarding their participation in daily life compared to those with early-onset forms (Kruitwagen-van Reenen et al., 2018). Increased fatigue and pain are more prevalent in the late-onset group, and motor impairment, exhaustion and depressive symptoms are key factors influencing participation. The emotional toll on parents is profound, particularly in SMA types I and II, where early death is a devastating reality (Higgs et al., 2016). Parents of surviving children and adolescents describe their experience as a lifelong challenge characterized by anticipatory grief, feelings of helplessness, and repeated emotional crises. These experiences are similar to those seen in people with other progressive, life-limiting conditions (Carlson & Brumback, 1983). Following a diagnosis of SMA types I or II, parents often begin preparing for the end of their child’s life, making decisions about how to spend their limited time together and how to say goodbye. Grief typically begins at the point of diagnosis and continues throughout the child’s life and beyond. Feelings of guilt relating to genetic transmission may also arise (Higgs et al., 2016). Accordingly, psychological and psychosocial support within a multidisciplinary framework is essential for affected families. Many parents report shifting their focus from seeking a cure to ensuring that their child’s life and death are meaningful. Despite the emotional intensity of this period, most parents in Higgs et al. (2016) described their child’s death as peaceful. Similarly, adults with SMA face psychological distress as functional decline, loss of independence and the perceived proximity of death become more pronounced (Ho et al., 2016; Kruitwagen-van Reenen et al., 2018; Lamb & Peden, 2008; Qian et al., 2015). In addition to the intensive daily demands of caregiving, parents of children with SMA often must invest a great deal of time and energy in navigating fragmented medical, psychosocial, legislative and financial support systems. Those caring for children with the more severe types I and II face additional challenges, such as complex treatment decisions, ethical dilemmas and palliative care considerations (Agosto et al., 2021; Hully et al., 2020). These parents are at an increased risk of experiencing psychological distress, fatigue and a diminished quality of life due to the burden of caregiving and limited resources (Dellve et al., 2006; Remedios et al., 2015), which often leaves them with little capacity for activities outside of caregiving. SMA presents a multifaceted challenge, with clinical manifestations and psychosocial impacts varying widely across individuals and families (Zerres & Rudnik-Schöneborn, 1995). Numerous studies have documented reduced quality of life among individuals with SMA and their families (Qian et al., 2015; von Gontard et al., 2002; Kočová et al., 2014). The progressive nature of the condition intensifies psychological distress, as patients and caregivers often describe the painful experience of anticipating functional decline while striving to preserve existing abilities. This uncertainty surrounding SMA progression significantly complicates coping and adaptation (Qian et al., 2015). Anticipatory grief can help families to gradually accept the impending death of a loved one, prepare for separation and adjust to life afterwards (Gross et al., 2012). However, for families of children with SMA, adolescence may paradoxically signal proximity to an early death (Yang et al., 2016). People with SMA often describe their experience as being characterized by suffering, which is not limited to severe subtypes, but reflects the challenges they face across the spectrum (Boardman, 2014). There is a broad consensus on the importance of integrating palliative care into the management of progressive neuromuscular conditions, including SMA (Ho & Straatman, 2013). Access to palliative care is generally adequate for families of children with SMA, primarily type I, focusing on respiratory support, nutrition, pain management, comfort and advanced care planning and end-of-life decision-making (Hully et al., 2020). Spirituality, which is often defined as the search for meaning or, in Christian contexts, as a relationship with God, can be an important coping mechanism (Woods & Ironson, 1999). Digital platforms, such as blogs and online communities, provide spaces where individuals with SMA can share personal stories, process challenging experiences, and find understanding and connection (Siuda & Pluta, 2020).
The death of a loved one can has a profound effect on the mental health and functioning of surviving family members. Severe grief in adulthood, characterized by intense emotional pain, longing and difficulty accepting the loss, has been linked to increased rates of depression and anxiety, the effects of which may persist for years (Thimm et al., 2020). Although grief affects all family members, existing research has primarily focused on parental bereavement following the death of a child. Respecting patient autonomy and clearly communicating the diagnosis and prognosis of progressive conditions such as SMA is crucial in fostering trust between clinicians, patients and caregivers. Such practices influence mental health outcomes and treatment decisions, helping to mitigate anxiety and distress with both immediate and long-term implications (Goodwin et al., 2015; Mendes et al., 2016; Verhaart et al., 2017; You et al., 2015). Open communication and transdisciplinary collaboration across specialties are essential to ensure coordinated, patient- and family-centered care (Murrell et al., 2018). When supporting families facing advanced stages of SMA, professionals from various disciplines must navigate the delicate balance between fostering hope and acknowledging the reality of impending death. Implementing specialized pediatric palliative care requires coordinated collaboration among stakeholders to optimize quality of life for patients and their families while respecting their preferences regarding treatment and end-of-life care (Hjorth et al., 2018; Lövgren et al., 2016). While disease-modifying therapies have altered the traditional course of SMA, they are not curative and are mainly used to delay progression (Xiao et al., 2023; Ch’ng et al., 2022).
Conclusions
Advancements in supportive care and the development of disease-modifying therapies have transformed the therapeutic landscape of SMA, greatly improving survival rates and life expectancy. An increasing number of individuals with SMA type II, as well as types III and IV, are reaching adulthood, largely thanks to enhanced medical interventions such as respiratory support.
However, SMA remains a progressive and life-limiting condition that requires long-term multidisciplinary medical and psychosocial care. This support is vital for preserving functional mobility, promoting independence and maintaining the highest possible quality of life. Understanding the lived experience of individuals with SMA is crucial. For many, ‘normalizing life’ means participating in everyday activities such as attending school, working, enjoying leisure activities, and contributing to family life, thus affirming their role as respected members of society. However, the most profound challenge lies in adapting to the ongoing progression of the disease. Everyone with SMA experiences a unique journey, which has a different impact on them and their families. The burden of care, emotional distress and psychosocial strain are daily realities for patients and carers alike. Although future treatments show promise, there is still a significant lack of published research on the lived experience of SMA, particularly regarding disease progression and end-of-life care. Further qualitative studies are urgently needed to address these unmet needs and inform more holistic, patient-centered approaches.
Review Limitations
This review was conducted using databases accessible through institutional licensing. Therefore, some relevant studies may have been excluded. Only English-language publications were considered, potentially excluding pertinent research published in other languages. As this is a qualitative review, the findings reflect the authors’ interpretations and understanding of the literature. Researchers with different backgrounds, methodologies or perspectives may reach different conclusions. It is also important to note that none of the authors are parents of children with progressive neuromuscular conditions, nor do they work directly with individuals affected by SMA or their families.
Footnotes
Authors’ Contribution
Conception and design (JCH, MH); data collection (JCH, MH); data analysis and interpretation (JCH, MH); drafting of the manuscript (JCH); critical revision of the manuscript (JCH, MH); final approval of the manuscript (JCH, MH). Both authors read and approved the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Psychosocial and Special Educational Needs of Children and Adults with Life-limiting Neurodegenerative Diseases and their Families (IGA_PdF_2024_018) implemented at the Faculty of Education, Palacký University Olomouc, Czech Republic.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.