
Abstract
In many human diseases, the molecular pathophysiological mechanisms are not understood, which makes the development and testing of new therapeutic approaches difficult. The generation and characterization of animal models such as mice, rats, fruit flies, worms or fish offers the possibility for in detail studies of a disease’s development, its course and potential therapies in an organismal context, which considerably minimizes the risk of therapeutic side effects for patients. Nevertheless, due to the high numbers of experimental animals used in research worldwide, attempts to develop alternative test systems will help in reducing their count. In this regard, the cell culture system displays a suitable option due to its potential of delivering nearly unlimited material and the good opportunities for high-throughput studies such as drug testing. Here, we describe a quick and simple method to isolate and cultivate vital fibroblast-like cells from embryos and adults of two popular teleost model organisms, the Japanese rice fish medaka (Oryzias latipes) and the zebrafish (Danio rerio).
Introduction
Expanding the scientific knowledge through basic biological research helps understanding how vital processes take place in the organism. Among others, these include the process of fertilization, development, growth, motion, sensory perception or aging. Biomedical science focuses on how pathophysiological mechanisms result from diseases and how these can be prevented or treated. The acquired knowledge could then be used for the benefit of patients.
It is undisputed that animal testing has already produced great benefits for humanity, including among others the discovery of the blood group system, insulin, penicillin or vitamins as well as the development of the electrocardiogram, cardiac catheterization, computer tomography or new techniques for organ transplantations in humans.1–5 Animal experiments are also indispensable for the development of new therapeutic approaches and the testing of drugs, for example against cancer or currently against viral infections caused by, for example, the ‘Severe Acute Respiratory Syndrome CoronaVirus 2’ (SARS-CoV-2) which leads to the still raging coronavirus disease 2019 (COVID-19).6,7 In a short time, several animal models have been developed contributing to the understanding of the mechanism of infection and to uncover therapeutic approaches against the virus. 8 This shows that studies conducted, for example in invertebrates such as Drosophila melanogaster and Caenorhabditis elegans as well as vertebrates such as frogs (Xenopus laevis and X. tropicalis), chicken (Gallus gallus domesticus), mouse (Mus musculus) and rat (Rattus norvegicus) or teleost fish model systems such as zebrafish (Danio rerio), medaka (Oryzias latipes) and swordtails (Xiphophorus) remain essential for research purposes.
Especially in recent years, the generation of animal models for biomedical research are steadily increasing, in part fueled by the improved targeted genetic modification techniques such as TALEN, zinc-finger nucleases or the popular CRISPR/Cas tool box and its derivatives.9–11 For 2015, it was calculated that around 192 million animals were used for scientific purposes worldwide. 12 Due to this huge number there is no question that the quantity of research animals should be reduced whenever possible, by applying the 3-R rule (Reduce, Refine, Replace) in animal experiments. The 3-R rule was developed and formulated by W.M.S. Russell and R.L. Burch as early as 1959 and became a legal EU Directive in 2013. 13 In this context, ‘Reduce’ means reducing the number of animals required to a minimum, ‘Refine’ means optimizing the methods used and ‘Replace’ means replacing animal experiments with alternative methods. These include, for example in-vitro 2D and 3D culture systems and in-silico computer simulation systems. In spite of its limitations for the investigation of complex developmental processes or multi-cell interactions, cell culture can serve as a highly efficient alternative to animal experimentation whenever standardized and high-throughput analyses such as drug screening approaches are needed. It can thus help as a substitute for experimentation on live animals (Figure 1). Another possible value of these generated fibroblast could be preserving the diploid genome via cryopreservation, thawing and somatic nuclear transfer into enucleated eggs.14,15 This accelerated cloning of animals, and supports preservation of genetic resources. Here, we present a quick and robust method for the isolation and cultivation of fibroblast-like cells derived from fish embryos or fin clip biopsies of adult medaka and zebrafish, with standard cell culture equipment (Figure 2). The cultured cells can be passaged for a long time and can be applied in a wide range of biochemical, genetic, therapeutic studies as a nearly unlimited and clonal resource.
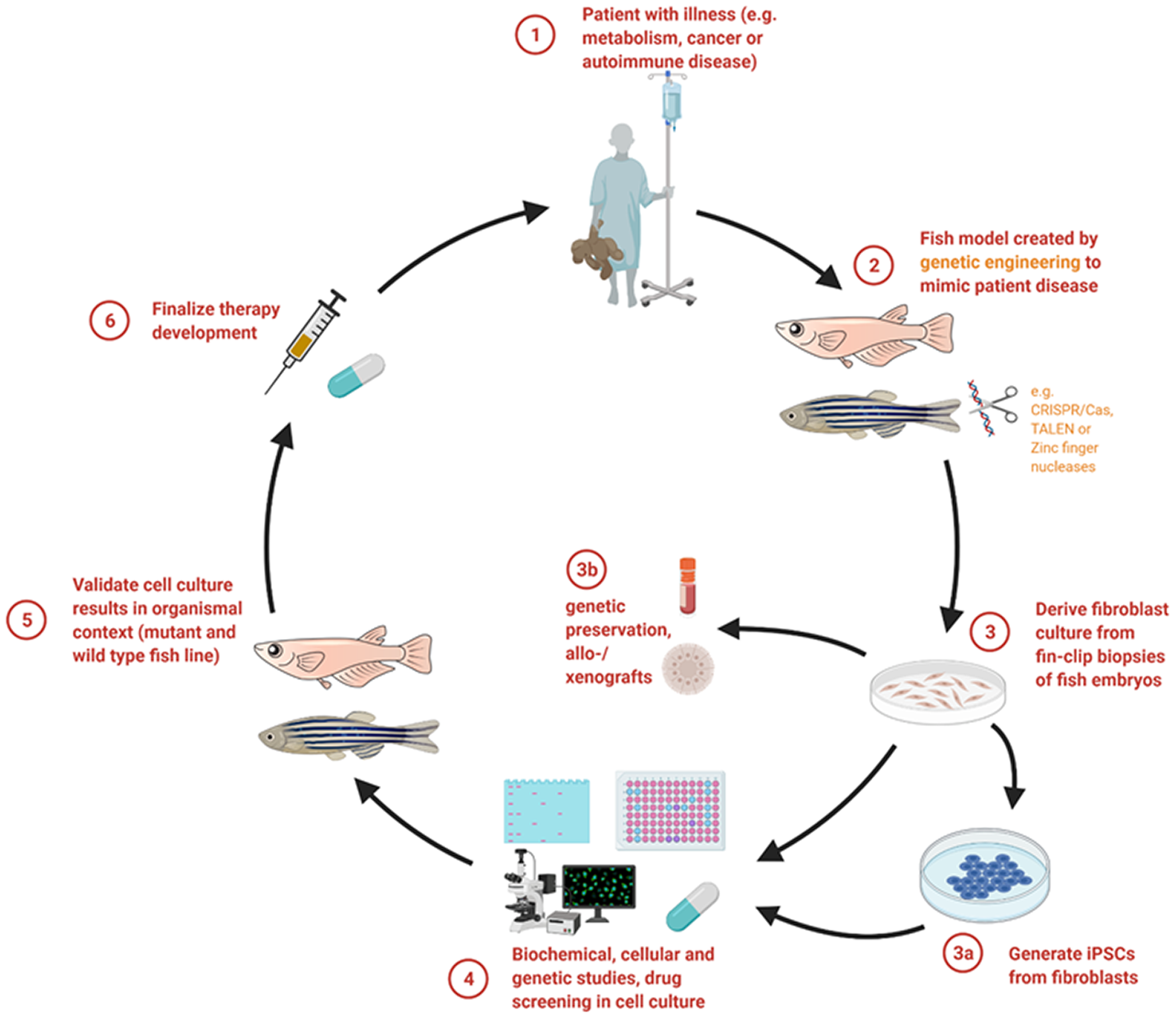
Synopsis of how personalized therapeutic approaches can be studied using a fish model in combination with cell culture analysis. Fish models mimicking a patient’s disease help to gain deeper insights into the pathophysiology and progression of the respective defect (steps 1 and 2). By using the cell culture system, already a wide range of initial experimental tests can be conducted (steps 3a, 3b and 4). Essential results gained by cellular models can then be applied and evaluated in the fish models (step 5) in order to finalize a treatment concept for patients (step 6). Created with BioRender.com.
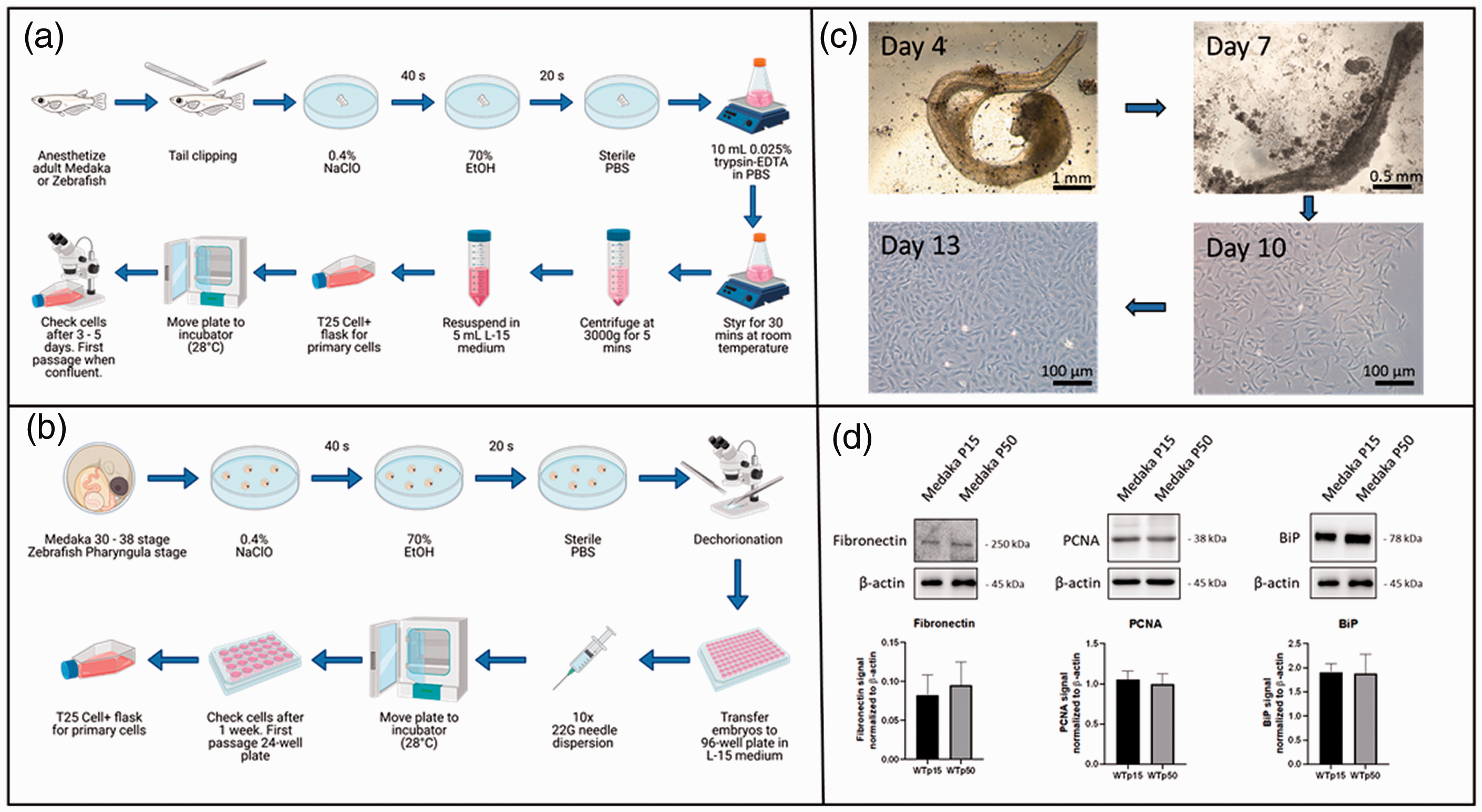
Cultivation of fibroblast-like cells derived from fish fins or embryos and biochemical analysis. (a) Flowchart for obtaining fibroblast-like cells from fish fins. (b) Flowchart for obtaining fibroblast-like cells from fish embryos. More information concerning the isolation and cultivation can be found in the methods section. Flowcharts were created with BioRender.com. (c) Microscopy images during the process of culturing cells derived from embryos (here from medaka). (d) Western blot analysis of fibroblast-like cells isolated from medaka embryos. The expression of fibronectin, proliferating cell nuclear antigen (PCNA) and immunoglobulin heavy chain-binding protein (BiP) revealed no side effects like cell degeneration even after 50 passages.
Animals, materials and methods
Animal husbandry
Medaka (O. latipes) Cab strain used in this study were kept as closed stocks in accordance to Tierschutzgesetz §11, Abs. 1, Nr. 1 and with European Union animal welfare guidelines. Medaka were maintained in a constant recirculating system with filters at 28°C on a 14 h light/10 h dark cycle, whereas wildtype zebrafish (D. rerio) were kept at 26 to 27°C in the same light and dark cycle. Cages used follow European regulations concerning space requirement and stocking density. The environmental conditions are monitored and routine health screening is performed continuously by animal attendants and veterinarians. Permissions by Regional council Karlsruhe, Germany: AZ35-9185.64/BH and AZ 35-9185.81/G-85/16.
Isolation and generation of fibroblast-like cells from fish fin clip biopsies
An adult medaka or zebrafish (>4 weeks) was anaesthetized in aquarium water supplemented with 168 mg/l tricaine (Sigma-Aldrich) for 10 min (Figure 2(a)). A maximum of 25% of the caudal fin was removed in a standard approach for genotyping and the fish was put in a basin with fresh aquarium water to recover from anaesthesia. The fin clip material was transferred for 20 s to a 100 mm Petri dish filled with sodium hypochlorite solution (0.4% NaClO in sterile water (w/v); Sigma-Aldrich), followed by soaking in ethanol (70% ethanol in sterile water (v/v); Sigma-Aldrich) for another 20 s in a third Petri dish. Afterwards, the fin clip biopsy was washed thoroughly in phosphate buffered saline (PBS; Sigma-Aldrich). With sterilized forceps the fin clip was transferred to a 50 ml beaker. For cell separation, 10 ml 0.025% trypsin-ethylenediaminetetraacetic acid (EDTA) in PBS was added and stirred with a magnetic stirrer (250 rpm; Heidolph) at room temperature until the fin was completely disassociated (max. 30 min). Next, the cell dispersion was centrifuged at room temperature for 5 min at 3000 g. All further procedures were carried out under sterile conditions in a laminar flow cabinet. The supernatant was carefully removed without disturbing the cell pellet. Next, the pellet was cautiously re-suspended in 5 ml Leibovitz’s L-15 medium (Gibco, Thermo Fisher) supplemented with 20% high quality fetal calf serum (FCS; PanSera), 1% penicillin/streptomycin (Gibco, Thermo Fisher) and 10 mM HEPES (Gibco, Thermo Fisher). To seed, the medium including the cells was transferred into a T25 cell flask assigned for primary adherent cells (Sarstedt) and was maintained at 28°C in a humidity-controlled cell culture incubator without disturbing for 48 h to give the cells enough time to settle and restart growing. After this incubation, the medium was carefully removed and discarded. Cells were washed twice with 3 ml PBS and were supplemented with 5 ml of fresh L15 medium with supplements. The medium exchange procedure was carried out until confluency of the cells (about 1 week). Passaging was performed by discarding the medium and washing the cells with 3 ml PBS. After removal of PBS, detachment of cells was achieved with 1 ml 1X accutase (Sigma-Aldrich) instead of trypsin-EDTA due to a better reattachment of cells afterwards. Incubation of the accutase-treated cells was continued at room temperature until they completely detached (about 10 min, check under microscope). Four ml L-15 medium were added to the flask with accutase and cells. Still existing cell conglomerates were detached by carefully pipetting up and down (avoid formation of foam). Dilutions of 1:2 to 1:5 were transferred into new T75 or T25 cell flasks for adherent cells (Sarstedt) for passaging.
Isolation and cultivation of fibroblast-like cells from fish embryos
For the isolation of fibroblast-like cells derived from embryos, 10 medaka embryos (stage 30–38 16 ; 3.5–8 days post-fertilization at 26°C) or zebrafish embryos (pharyngula stage 17 ; 24–48 h post-fertilization at 28.5°C) were transferred into a 100 mm Petri dish filled with 0.4% NaClO (Sigma-Aldrich) for 30 s (Figure 2(b)). Next, the embryos were placed in a second dish filled with 70% ethanol and incubated for 20 sec. Finally, they were transferred to a third Petri dish filled with sterile PBS for thorough washing.
Under a stereomicroscope, preferably in a laminar flow bench, dechorionization (removal of egg envelope, chorion) was carried out. The chorion was punctured with a G26 needle and ripped apart with sterilized forceps. Please note that the commonly used two-step protease treatment for dechorionization (pronase and hatching enzyme) is not suitable here, as a sterile environment is needed. In case that deyolking has not occurred spontaneously during dechorionization, the yolk sac was removed from the embryo with help of a sterile pipette fitted with a 200 µl infiltered tip. The pipette tip was attached to the yolk sac and mild pressure was applied. Hereby the yolk sack burst and all its material was sucked off afterwards. Next, 150 µl of L-15 medium supplemented with 20% high quality fetal calf serum (FCS; PanSera), 1% penicillin/streptomycin (Gibco, Thermo Fisher) and 10 mM HEPES (Gibco, Thermo Fisher) were added per well of a 96-well plate. One embryo (without chorion and yolk sac) was transferred per well with sterilized forceps. Disintegration of each embryo was accomplished by pipetting carefully up and down 10 times with a sterile G22 needle and syringe by avoiding formation of foam. After closing the lid, the 96-well plate was placed in a humidity-controlled cell culture incubator. Incubation was carried out at 28°C for 7 days without disturbing. When the cells reached confluency, cells were washed three times with PBS and passaged to a 24-well plate. For detachment of cells 1X accutase (Sigma-Aldrich) was used. Incubation of the accutase-treated cells was maintained at room temperature until they detached (about 10 min, checked under microscope). Next, 100 µl of L-15 medium with supplements was added and the cell suspension (without residual embryonic conglomerates) was transferred to a new 24-well plate for adherent cells. When confluency was reached, cells were transferred to a 6-well plate or alternatively T25 Cell+ flask for adherent cells (Figure 2(c)).
Verification of cultivated cells by Western blotting
Fibroblast-like cells from medaka embryos of passages 1, 15 and 50 were washed with ice-cold PBS and harvested by cell scraping in RIPA buffer (Thermo Fisher). Cells were solubilized by passing the sample 10 times through a G22 needle and syringe followed by ultra-sonic homogenization in an ice-water bath. Next, fibroblast suspension was centrifuged at different speeds. For isolation of the nuclear fraction, centrifugation was carried out at 1000 g for 10 min. To obtain the cell membrane and ER fractions, centrifugation was performed at 100,000 g for 1 h at 4°C and the protein concentrations of the respective pellet fractions were analysed by DC protein assay kit (BioRad).
To verify that fibroblast-like cells were isolated and cultivated, expression of fibronectin was analysed by Western blotting with 10 μg protein derived from the cell membrane pellet fraction. Samples were mixed with 6X Laemmli buffer (375 mM Tris–HCl, pH 6.8, 6% SDS, 48% glycerol, 9% 2‐mercaptoethanol, 0.03% bromophenol blue) and denatured for 5 min at 95°C. Extracts were analysed on Mini-PROTEAN®TGX™ gradient Gels 4–15% (Bio-Rad, Munich) and blotted onto a nitrocellulose membrane (GE Healthcare) by semi‐dry electrophoretic transfer. The membrane was blocked for 2 h at room temperature with 5% milk powder in PBS‐T (0.1% Tween in phosphate buffered saline (PBS)). After blocking, the membrane was washed with PBS-T and incubated over night with the primary antibody fibronectin (rabbit, anti-human, 15613-1-AP; Proteintech) in a dilution of 1:1,000 in PBS-T at 4°C. Washing steps with PBS-T were conducted three times and the membranes were incubated for 1 h at room temperature with the secondary antibody IgG conjugated with horseradish peroxidase (HRP) (goat, anti-rabbit; Dianova) diluted 1:10,000 in PBS-T. Protein signals were detected by light emission with Pierce™ ECL Western blot analysis substrate. Blots were stripped for 15 min in 10% acetic acid and blocked again with 5% milk powder in PBS-T. For quantification, the primary antibody β‐actin (mouse, anti-human; Sigma-Aldrich) and the secondary antibody IgG‐HRP (goat, anti-mouse, Santa Cruz) in dilutions of 1:1000 and 1:10,000 in PBS-T were used, respectively.
Cellular proliferation and determination of ER stress were addressed by expression analysis of the proliferating cell nuclear antigen (PCNA, also named cyclin) and ‘Immunoglobulin heavy chain-binding protein’ (BiP, also named GRP78 or HSPA5. For PCNA and BiP, 10 µg of the cellular pellet and of the cell membrane and ER fraction pellet were used per lane on a 10% SDS-PAGE. Further procedure was as described above. As primary antibodies PCNA (rabbit, anti-human, 10205-2-AP; Proteintech) and BiP (rabbit, anti-human, 11587-1-AP; Proteintech) were used in dilutions of 1:1000 for each antibody in PBS-T. As secondary antibody IgG‐HRP (goat, anti-rabbit, Santa Cruz) in a dilution of 1:10,000 in PBS-T was used.
To verify whether cells of the passage 1 display multipotency, expression of ‘sex determining region Y (SRY)-box 2’ (SOX2) was analyzed. Protein extraction and Western blotting was performed as described above. The SOX2 antibody (rabbit, anti-human, ab97959; Abcam) was used in a dilution of 1:1000, the secondary antibody IgG‐HRP (goat, anti-rabbit; Santa Cruz) was used in a dilution of 1:10,000 in PBS-T. In this course we also checked for cardiomyocytes. We used α-Actinin (ACTN2) as a marker as it is specific for a-cardiac muscle actinins. The primary antibody ACTN2 (mouse, anti-rabbit, A7811; Merck) and the secondary antibody IgG-HRP (goat, anti-mouse, Santa Cruz) were used in the same dilutions as mentioned above.
Results
This study was conducted with the goal to reduce animal experiments by using the cell culture system. With the method presented here (Figure 2(a) and (b)), it was possible to generate large amounts of fibroblast-like cells for a variety of experiments within a short period of time. The morphology of the cells derived from embryos or fin clip biopsies resembled the shape of fibroblasts (Figure 2(c)). The average time until a confluent T75 cell culture bottle (c. 3 million–5 million cells) was available, was 11–14 days when started from embryos and 5–7 days when started from fin clips.
We did not observe any temporal differences in the cultivation of cells derived from zebrafish or medaka.
Nevertheless, when fibroblasts were isolated from embryos, some pulsating cells were found, which we assume to be an accumulation of cardiomyocytes. In 10 preparations from embryos, the pulsating cell accumulations were observed on average in 2–3 wells. In the course of further cultivation in L15 medium, these disappeared within the next two passages, and a homogeneous cell image was detectable under the microscope. The presence of cardiomyocytes in passage 1 was confirmed by Western blot analysis of ACTN2 (Figure 3(a)). A band for ACNT2 (0.68 arbitrary unit (a.u.) ± 0.07) was only detectable for passage 1 whereas passage 15 and 50 showed no signal at all. Expression analysis of fibronectin (0.08 a.u. ± 0.03 to 0.09 a.u. ± 0.03) (Figure 2(e)) demonstrated that after 15 as well as after 50 passages, the cell culture flasks mainly contained fibroblast cells.
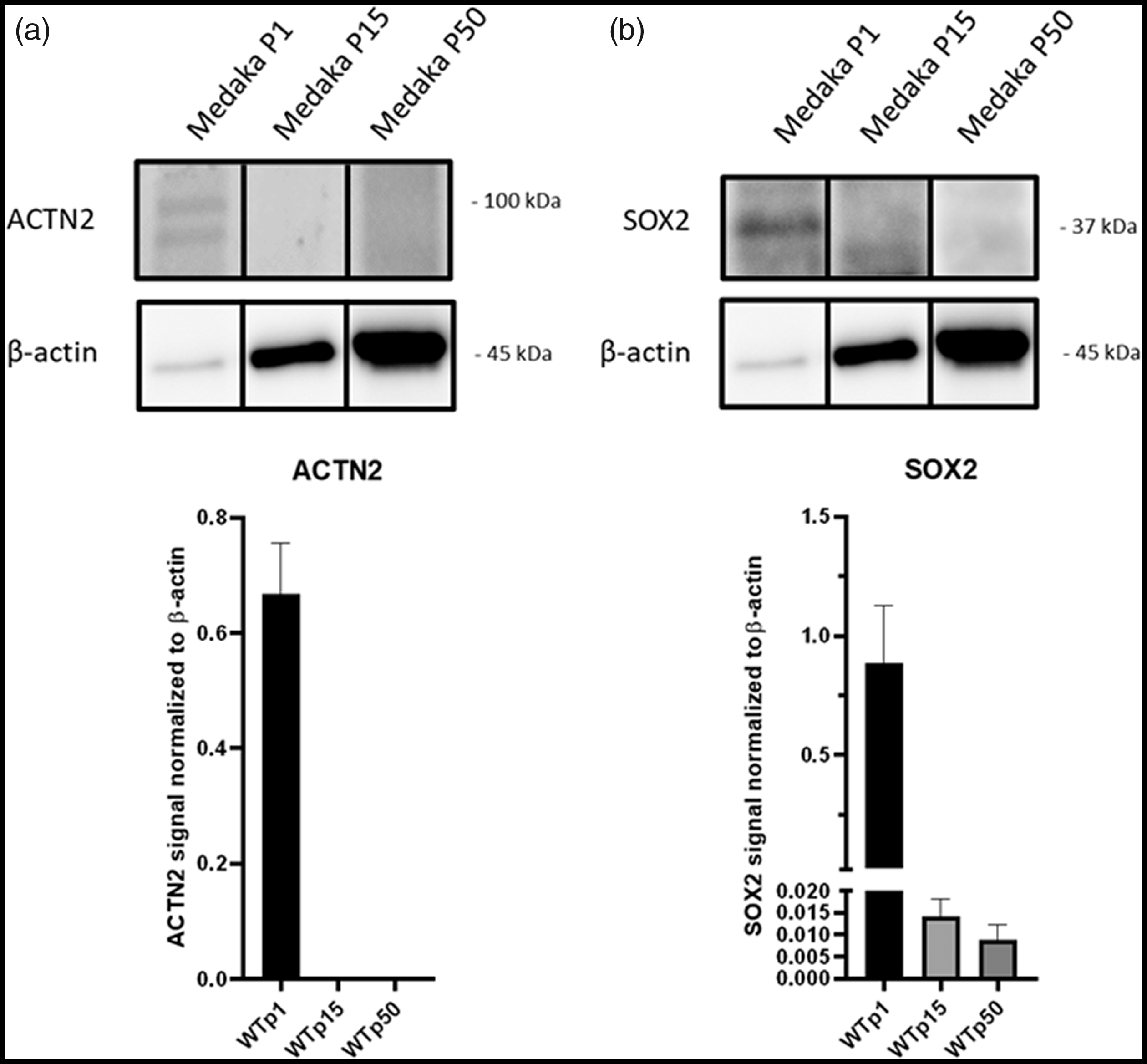
Expression analysis of sex determining region Y (SRY)-box 2 (SOX2). Protein material for Western blotting was derived from cell passages 1, 15 and 50 of Medaka embryos (stage 30–38). (a) α-Actinin (ACTN2) and (b) SOX2 expression was normalized to β-actin. ACTN2 protein level was only visible in passage 1 and SOX2 protein level was highly expressed in passage 1, whereas in passages 15 and 50 nearly no expression was detectable, indicating presence of cardiomyocytes and undifferentiated embryonic stem cells in passage 1.
Cells that were isolated from embryos and those that were gained and cultivated from fin clip biopsies showed a high degree of vitality and no morphological abnormalities even after 50 passages. The comparison of the expression of PCNA of cells from passage 15 and cells from passage 50 showed comparable protein levels (1.065 a.u. ± 0.11 to 0.10 a.u. ± 0.13) as well (Figure 2(e)). Furthermore, the increasing number of passages did not have a negative effect on the cellular stress response which showed comparable signal strengths for BiP (GRP78) at both passages investigated (passage 15: 1.91 a.u. ± 0.17 to passage 50: 1.88 a.u. ± 0.39) (Figure 2(e)).
To elucidate, whether in passages 1, 15 and 50 of embryos multipotential stem cells were present, Western blotting of SOX2 was carried out. High expression of SOX2 was revealed in passage 1 (0.886 a.u. ± 0.194), whereas in passages 15 and 50 only weak expression was detected (0.014 a.u. ± 0.003 and 0.009 a.u. ± 0.003, respectively) (Figure 3(b)).
Discussion
The rigorous tightening of animal experimentation law, the legally required biometric planning and the expansion of the responsible control authorities in combination with the increased vigilance of private and public third-party funds have already effectively reduced the amount of animal experimentation and number of animals used in biomedical research. In addition, new measures to protect animals and reduce their exposure have been constantly researched and implemented for years (anesthesia, analgesia, strict criteria for stopping experiments, self-limitation of the sciences). In addition, there are new technologies such as multi-organ chips or computer simulations with which, for example, predictions can be made about the effect of a treatment substance on humans. 18 However, in 2015 over 190 million animals were still used in research and medicine worldwide. 12 Although a complete abolition of animal experiments will not be possible without risking patient safety and research quality, certain cell and tissue specific questions may be addressed in an ex-vivo context. Such attempts will significantly decrease the total number of experimental animals required.
With the method presented here, fibroblast-like cells can easily and robustly be isolated from embryonic stages or as a byproduct of material derived for genotyping during husbandry routine (fin clip biopsy) of medaka and zebrafish. In this way, fewer animals need to be sacrificed but rather unlimited recourses are generated for a plethora of investigations.
In comparison to many of the previously published protocols for the establishment of fibroblasts from fish material, the method presented here offers the decisive advantage that fibroblasts are obtained from one individual (single embryo or single caudal fin of an adult fish).19,20 The cell lines subsequently obtained thus display with a homogeneous genetic background.
It cannot be ruled out that during the isolation, the fibroblasts were contaminated with other cell types. For analysis, we concentrated on the cells derived from medaka embryos and checked the expression of fibronectin as one of the major components of the extracellular matrix that is secreted by several cells, but foremost by fibroblasts. Both the morphology of the cells and the result of the Western blot on fibronectin indicated the presence of fibroblasts or at least fibroblast-like cells in the Petri dishes. Notably, in the first two passages accumulation of pulsating cells were observed which indicated an initial contamination potentially by cardiomyocytes. This assumption is supported by the expression of ACTN2 as a marker for cardiomyocytes only found in cells derived from passage 1 but not passages 15 and 50. 21
In particular, we also found high expression of SOX2 in cells of passage 1. This protein is essential for the maintenance of self-renewal of undifferentiated embryonic stem cells and its expression showed that there are still numerous progenitor cells to organs in early cell passages obtained from embryos. 22 Basically, our method can therefore offer the possibility to enrich for other cell types such as heart cells, neurons or hepatocytes by using selective and special media over several passages instead of L-15 medium. 23 The generated fibroblasts could also serve for genetic preservation as well as allo- and xenografts (Figure 1).
We found that the cultivation of fibroblasts from fin clip biopsies was naturally associated with a reduced risk of contamination by cells of other organs. An advantage of using embryos for the cultivation of fibroblasts may be the generation of other cell types such as iPSCs, which could also be further differentiated. All this is consistent with and in support of the goals of the 3Rs.
The isolation and cultivation of fish cells turned out to be simple and comparable to that of human or murine skin fibroblasts. A major difference to the mammalian fibroblasts, however, is that the fish cells seem to degenerate much more slowly. Degeneration can usually be seen in terms of a change in cellular morphology, decreased proliferation, or increased cell stress, among other things. Notably, even after 50 passages, we did not observe a change in the macroscopic structure of the cultivated cells. PCNA is a nuclear nonhistone protein that is necessary for DNA synthesis and is an accessory protein for DNA polymerase alpha. Quiescent and senescent cells have very low levels of this protein which is widely used as a marker for proliferation. 24 We found comparable strong signals for PCNA in passages 15 and 50, which provided evidence that cell proliferation was still present. BiP displays an endoplasmic reticulum (ER) stress marker, as it is a major chaperone protein critical for protein control of the ER, as well as controlling the activation of the ER transmembrane signaling molecules. 25 Since we detected comparable signals for BiP protein expression at passages 15 and 50, we conclude that the fibroblast-like cells cultivated by this protocol showed consistent cellular fitness, even after long-time cultivation. Although the scope of the biochemical analysis was on medaka, the cultivated fibroblast derived from zebrafish showed similar cellular morphology and growth, suggesting a high comparability of zebrafish and medaka cells.
These cells thus represent potent source of renewable experimental material, such as DNA, RNA, proteins, lipids, sugars, amino acids, etc. for a multiplicity of genetic, biochemical as well as metabolic analyses. Those studies can be carried out just as easily as tests for effectiveness and toxicity of a substance or even a holistic therapeutic approach. By such preliminary investigations in cell culture, already significant gains in knowledge can be achieved and the number of test animals in biomedical research can be effectively reduced.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this study was supported by the Deutsche Forschungsgemeinschaft (FOR2509: to C. Thiel: TH1461/7‐2 and J. Wittbrodt: WI1824/9-1).
Research ethics statement
All experiments were performed before the embryos’ free-feeding stage or with fin tissue which was taken from adult fish in the course of genotyping and so did not fall under animal experimentation law according to the EU Animal Protection Directive 2010/63/EU.
Contributions
LB planned and conducted experiments, contributed to the writing of the manuscript; SG, AH conducted experiments; TT, JW collected data and contributed to the interpretation of the results; CT, planning of experiments, interpretation of data and writing of the manuscript.
Data availability statement
The data that support the findings of this study are available from the corresponding author, [CT], upon reasonable request.