
Abstract
Directive 2010/63/EU introduced requirements for the classification of the severity of procedures to be applied during the project authorisation process to use animals in scientific procedures and also to report actual severity experienced by each animal used in such procedures. These requirements offer opportunities during the design, conduct and reporting of procedures to consider the adverse effects of procedures and how these can be reduced to minimize the welfare consequences for the animals. Better recording and reporting of adverse effects should also help in highlighting priorities for refinement of future similar procedures and benchmarking good practice. Reporting of actual severity should help inform the public of the relative severity of different areas of scientific research and, over time, may show trends regarding refinement. Consistency of assignment of severity categories across Member States is a key requirement, particularly if re-use is considered, or the safeguard clause is to be invoked. The examples of severity classification given in Annex VIII are limited in number, and have little descriptive power to aid assignment. Additionally, the examples given often relate to the procedure and do not attempt to assess the outcome, such as adverse effects that may occur. The aim of this report is to deliver guidance on the assignment of severity, both prospectively and at the end of a procedure. A number of animal models, in current use, have been used to illustrate the severity assessment process from inception of the project, through monitoring during the course of the procedure to the final assessment of actual severity at the end of the procedure (Appendix 1).
Introduction
Implementation of Directive 2010/63/EU has imposed additional requirements related to the severity of procedures carried out on animals for scientific purposes. Procedures need to be assigned a severity classification prospectively and the actual severity experienced by each animal during the course of a procedure has to be determined and reported in the statistical information made publicly available annually (Commission Implementing Decision 2012/707/EU, as amended by 2014/11/EU).1,2
Good project planning is necessary to determine a suitable prospective severity classification and to develop appropriate observational monitoring and assessment criteria and humane end-points, tailored to the study.3,4 Sufficiently trained and competent staff are an absolute requirement to assess animal welfare during the course of the study.
There needs to be an observational strategy and a common recording system that captures all the necessary data in a consistent format to facilitate continued application of refinement and enable an assessment of actual severity to be made. This paper provides the rationale of why such assessment is necessary and who is responsible for it. It then develops several real examples of animal procedures of how to do so and how this then allows evaluation of the actual severity score for each individual animal that has gone through the procedures.
Annex VIII of the Directive has included some additional guidance on prospective severity classification to help achieve some common interpretation of the Directive’s intentions.
The examples give little information on how the severity classification was derived and give a mix of simple single step procedures, such as short term restraint in a metabolic cage and more complex procedures which comprise multiple steps such as organ transplantation requiring anaesthesia, surgery and management of organ rejection.
Although the assignment criteria (set out in Section II of Annex VIII) indicate that each study needs to consider a range of factors before a classification is made, many within the scientific community have raised concerns that the examples provided in Annex VIII contain insufficient information to satisfactorily explain the rationale for the severity classification and that no examples are provided for some important areas of research, such as, for example, pain and arthritis. Without additional explanation, there are likely to be considerable differences in the assignation of severity, which may ultimately give misleading information on animal use, and, perhaps of greater concern, result in inappropriate re-use of animals. A further potential concern related to animal models of pain is that it is possible for the same model to be categorised across at least two severity classifications, depending on the refinements in the procedure. In addition to the application of early end-points, the degree of amelioration of pain, distress and suffering is a major factor.
During 2012, members of the FELASA/ECLAM/ESLAV Working Group contributed to discussions at a meeting arranged by the European Commission on severity classification, and using material developed by this group, assisted in the development of some additional guidance and a few examples of severity classification which were endorsed at a National Contact Point (NCP) meeting and can be found at the EC website. 5
A main purpose of this joint FELASA/ECLAM/ESLAV report is to provide additional information and guidance on prospective severity classification and assessment of severity experienced by the animals during the course of a procedure (actual severity), through a number of further illustrative examples from different fields of research drawing on existing systems, providing examples of different severities and expanding on the examples provided in Annex VIII. A number of ‘severe’ models have been deliberately included to illustrate areas of animal use in scientific work not included in Annex VIII and to facilitate sharing and dissemination of good practice.
Models have been chosen which, at the time of preparation of this report, were in use in fundamental and applied research, together with some examples used in safety evaluation. The examples include some information on how severity can be reduced through application of refinement strategies. Additional suggestions for refinement were incorporated following review by the parent organisations.
Although the illustrative examples are representative of current practices, the principle remains that whenever the use of any animal model is proposed, each component of the study should be reviewed and challenged where appropriate to ensure that all 3R opportunities are applied.
With these additional requirements in 2010/63/EU, it is important for all involved in the use of animals, including those responsible for project evaluation, to develop and agree a common understanding of and approach to ‘severity classification’ in order to promote a ‘level playing field’ within the European research community. This should thus ensure a consistent reporting of the severity experienced by the animals during the procedures in the statistical returns on animal use.
The regulatory framework
The new Directive 2010/63/EU on the protection of animals used for scientific purposes was approved on 22 September 2010 and took full effect in Member States on 1 January 2013.
As with Directive 86/609/EEC, the new Directive requires that experiments are designed to cause the least pain, suffering, distress or lasting harm.
All scientific procedures will be conducted under a project authorisation approved in each Member State (MS) by the Competent Authority (CA). All those applying for project authorisation will need to include an estimate of the likely severity of each procedure. These severity estimates will be considered by the CA during the project evaluation process undertaken, before a decision on project authorisation is made. Having considered the information provided in the application, the CA will assign a severity classification to each procedure (Article 38).
The actual severity experienced by each animal during each individual procedure will be reported by each MS (Article 54(2)) annually, in the year in which the procedure is completed.
Furthermore, the
Article 3 defines a procedure as ‘any use, invasive or non-invasive, of an animal for experimental or other scientific purposes, with known or unknown outcome, or educational purposes, which may cause the animal a level of pain, suffering, distress or lasting harm equivalent to, or higher than, that caused by the introduction of a needle in accordance with good veterinary practice’.
This defines a lower ‘threshold’ for a scientific procedure below which project authorisation will not be necessary. This definition gives an indication of the level of pain which could be considered as a ‘threshold’, but there is no information given on equivalent thresholds for suffering, distress or lasting harm.
Since the adoption of the Directive, EU guidance on severity assessment was developed and endorsed in 2012 and additional information to promote consistent reporting was included in a discussion paper from the NCP meeting in January 2016.6,7
Why do we need a severity classification system?
The inclusion of a severity classification system within the new Directive provides an opportunity to focus continuously on refinement from inception to completion of a procedure, improving the quality of science and animal welfare, and, by the inclusion of the actual severity experienced by each animal during a procedure in the Statistical Reports, providing greater transparency and promote improved public confidence in the use of animals in research. Over time, these publications may provide information on trends in refinement.
A number of European countries, including Finland, Germany, Ireland, The Netherlands, Poland, Sweden, Italy, Switzerland and the UK, and Australia, Canada and New Zealand have, for a number of years, had in place systems to categorise the severity of animal studies.
Many of the existing systems report prospectively, with the number of categories varying from 3 to 9. 8 None of the systems, however, use the combination of prospective, actual and cumulative suffering or the classifications included in the new Directive. Assignment of prospective classification and reporting of actual severity are necessary to enable comparison during retrospective review of a project, where such review is required.
The severity categories are defined in Annex VIII of the Directive as follows: The severity of a procedure shall be determined by the degree of pain, suffering, distress or lasting harm expected to be experienced by an individual animal during the course of the procedure. Non-recovery: Procedures, which are performed entirely under general anaesthesia from which the animal shall not recover consciousness shall be classified as non-recovery. Mild: Procedures on animals as a result of which the animals are likely to experience short term mild pain, suffering or distress, as well as procedures with no significant impairment of the wellbeing or general condition of the animals shall be classified as mild. Moderate: Procedures on animals as a result of which the animals are likely to experience short term moderate pain, suffering or distress, or long-lasting mild pain, suffering or distress as well as procedures that are likely to cause moderate impairment of the wellbeing or general condition of the animals shall be classified as moderate. Severe: Procedures on animals as a result of which the animals are likely to experience severe pain, suffering or distress, or long-lasting moderate pain, suffering or distress as well as procedures, that are likely to cause severe impairment of the wellbeing or general condition of the animals shall be classified as severe. Note: There is the possibility with exceptional and scientifically justifiable reasons for Member States to adopt a provisional measure to permit the use of a procedure involving severe, pain, suffering or distress that is likely to be long-lasting and cannot be ameliorated. Any such provisional measures must be considered and approved by an EU committee for such work to continue (Article 55).
An estimate of severity expected to be experienced by the animal has to be given for each scientific procedure. This requirement provides an opportunity during the design of the study to consider the application of the 3Rs and to ensure that the severity is reduced as far as possible within the scientific constraints of the study.9,10
This consideration of severity should therefore benefit animals by reducing suffering, and may also improve robustness of scientific design by giving opportunities to consider the effects of the procedures on, for example, physiology or behaviour where, for example, deteriorating health/welfare could affect outcomes, and ways by which such changes can be minimised to improve the quality and consistency of data.
The classification will furthermore help to define clear upper limits on animal suffering, and thus can assist in the implementation of humane end-points.
The 3Rs should continue to be reviewed as the project develops both by those directly involved in the use of animals and by the Animal Welfare Body (AWB – as detailed in Article 27 of the Directive).
When required, a retrospective assessment (RA) of a project gives a further opportunity to review the welfare costs/harms to the animals, to determine whether the objectives have been met, and to re-consider the appropriateness of the severity classification, prior to any future study.
Who determines the severity classification?
The application for a project authorisation by the user or the person responsible for the project requires that a
The CA which conducts the project evaluation (Article 38) shall include an ‘assessment and assignment of the classification of the severity of procedures’. The CA will consider expertise in relevant scientific areas, experimental design, laboratory animal science or wildlife veterinary practice and animal husbandry and care, as appropriate for the project proposal.
Prospective severity classification is assigned to the procedures by the CA during project evaluation, and this shall be based on the most severe effects likely to be experienced by an individual animal after all refinements have been applied.
The AWB (Articles 26 & 27) is required to follow the development and outcome of projects and to advise on opportunities for the application of the 3Rs within these projects.
The
The National Committees for the protection of animals used in scientific procedures (Article 49) are expected to promote and share best practices within the European Union. An important aspect of their role will be to promote consistency with regard to severity assessment.
Terminology
Some clarification and standardisation in terminology is necessary to ensure a common approach is taken to the assessment and assignment of severity classification.
The Technical Expert Working Group (TEWG) convened by the European Commission in 2003 to consider various aspects of the composition of the new Directive made several recommendations with regard to the terminology that should be used. These have not been directly transposed and the lack of further explanation in Annex VIII has contributed to further confusion on what aspects of the procedures (within a project) have to be assessed for severity. Directive 2010/63/EU – Article 3 ‘procedure’ means any use, invasive or non-invasive, of an animal for experimental or other scientific purposes, with known or unknown outcome, or educational purposes, which may cause the animal a level of pain, suffering, distress or lasting harm equivalent to, or higher than, that caused by the introduction of a needle according to good veterinary practice. This includes any course of action intended, or liable, to result in the birth or hatching of an animal or the creation and maintenance of a genetically modified animal line in any such condition, but excludes the killing of animals solely for the use of their organs or tissues; ‘project’ means a programme of work having a defined scientific objective and involving one or more procedures;
The recommendation from the TEWG Authorisation Sub-Group was to separate the definition of a project from an ‘experiment’ and recommended that the term ‘procedure’ should be used rather than ‘experiment’, to include both procedures with known outcomes (e.g. procedures concerned with the production of antibodies) and with unknown outcomes (e.g. a procedure conducted to test a hypothesis). This concept was included but the further recommended division was not. Technique: A technical act on one or more animals for an experimental or other scientific purpose and which may cause that animal or those animals pain, suffering, distress or lasting harm. Examples of technical acts would be gavage, injection, laparotomy, withholding of food/water. Procedure: A combination of one or more technical acts carried out on an animal for an experimental or other scientific purpose and which may cause that animal pain, suffering, distress or lasting harm. Project: A coherent programme of work aimed at meeting a defined scientific objective or objectives and involving a combination of one or more procedures.
At a NCP meeting in October 2011, the concept of a single and multiple-step procedure was preferred to the use of ‘technique’, as this term is not included in the Directive.
The term ‘procedure’ should, therefore, be used to describe the complete series of steps (techniques) that need to be applied to complete a particular experimental or other scientific purpose. Some procedures may include only a single step (technique) (e.g. withdrawal of blood from ‘normal’ animal to enable in vitro studies), but the majority will include a number of steps (techniques) (e.g. a vaccine challenge study could involve injection of vaccine, exposure to an infectious agent, and sampling or biopsy of tissues).
Annex VIII in the Directive provides a number of examples of types of procedures in the different severity categories, and includes a mix of single step procedures and multiple-step procedures.
To determine the severity of a procedure, consideration will need to be given to the contribution to the overall severity made by each step (technique) (and the consequences of each step) within a procedure. For example, when injecting a substance, consideration needs to be given on the impact of the injection itself, and also on any subsequent effects of the substance being injected.
Prospective severity classification
The final classification of a procedure will be determined by the most severe effects expected to be experienced by
Actual severity reporting
In contrast to prospective classification, the actual (highest) severity experienced by
There will therefore likely be differences in severity between prospective severity classification for the procedure and the actual severity reported for each of the animals used in the procedure.
‘Below threshold’ for regulation
Directive 2010/63/EU defines a ‘procedure’ as ‘an intervention which may cause the animal a level of pain, suffering, distress or lasting harm equivalent to, or higher than, that caused by the introduction of a needle according to good veterinary practice’.
Annex VIII gives some examples that fall below this threshold. These include minimal restraint of habituated animals, application of external telemetry devices and minor dietary manipulations, including variations in composition and availability, provided these are not expected to cause any adverse effects.
The Annex also notes that consideration needs to be given when a frequency or combination of ‘below threshold’ interventions may result in a cumulative effect which leads to the classification of the procedure as ‘mild or higher’.
For example, scientific investigations into novel husbandry practices which involve significant changes to cleaning frequency and disruption to social groups are likely to cause some suffering and distress and therefore would require project authorisation.
Severity assessment of procedures and the harm–benefit analysis of projects
The harm–benefit analysis that is required for project evaluation (Article 38) will take into consideration the likely impact of all animals used within the project, and therefore needs to take account of all potential harms to all animals.
In contrast, the severity classification of each procedure will give an indication of the limit of the suffering to a single animal used within the procedure.
It follows therefore that the information needed for project evaluation needs more detail on the welfare harms to all animals than that provided by a simple severity classification applied to the procedures contained within the project.
For example, in a vaccination challenge study, some animals (unvaccinated controls) may experience severe clinical disease, requiring an assignment of ‘severe’ to the procedure.
However, and in contrast, it would be expected that the majority of the animals given an effective vaccine are likely to experience no more than mild adverse effects.
This detailed understanding of the likely impact on all the animals to be used in a project is necessary to permit an informed harm–benefit analysis. 10
In the example above, where the project consists of developing a novel vaccine against a disease that is associated with high mortality, the harm–benefit analysis is likely to be in favour of the project. Of course, this would be dependent on consideration of many other factors, such as experience and implementation of the 3Rs in the procedures.
Planning of a procedure
The applicant should discuss the project proposal with the veterinarian (or suitably qualified expert where more appropriate), care staff and/or AWB to consider the procedures to be applied, the opportunities to apply the 3Rs, for example appropriate dosing and sampling strategies, and to agree appropriate monitoring/assessment criteria, interventions to minimise suffering and where applicable humane end-points.
All those involved in severity assessment should have a sound understanding of animal behaviour and welfare and of the indicators of poor welfare, pain and suffering in the species being used. 4
This process ensures that all personnel involved in the studies have an opportunity to contribute to the study design, and to ensure that all are aware of the potential adverse effects, the animal monitoring which will be in place and the methods to be implemented to minimise suffering.
Effective teamwork among all those involved is necessary to ensure consistent interpretation and minimisation of suffering compatible with the scientific objectives.
Training in severity assessment
Although the project leader will be responsible for returning the data on actual severity to the CA, often the assessment of actual severity will be undertaken by those directly involved in carrying out procedures and observing and caring for the animals.
Ensuring that all those involved have been appropriately trained and have a good knowledge of normal and abnormal behaviour in the species/strain being used is therefore essential.
How should severity be assessed?
Prospective severity classification of procedures
Many factors have to be taken into consideration in order to determine a suitable severity classification for a procedure.
Although assessment is largely subjective, as more scientific information becomes available, our understanding of how to recognise pain, suffering and distress improves, and it is therefore important to remain abreast of developments in this rapidly evolving field of research.
Some examples of severity classifications of procedures are included in Annex VIII of the Directive 2010/63/EU and in the endorsed EU Severity Assessment Framework.
When determining an appropriate severity classification, it is necessary to consider the impact on the animal of each step of the planned procedure:
What is being done to the animals?
What effect will this have on the animals?
How much suffering may it cause?
What interventions can be included to reduce the impact on the animals?
What is being done to the animals?
Consider all the steps involved in the procedure:
Changes to normal environment, husbandry and care practices Conditioning/training; handling and restraint Administrations/injections of substances – routes, volumes, frequency Sampling – what is being sampled, from where is the sample being taken, how much and how frequently are the samples being taken Surgical and other invasive interventions Use of anaesthesia – local, general, regional and/or analgesia Duration of study In the case of work in the wild – method and frequency of capture, accidental capture of non-target species, temporary housing, etc.
What effect will these interventions have on the animals?
Changes to the environment, husbandry and care practices may initially impact animal welfare and cause changes in behaviour (e.g. increased aggression), but habituation of the animals will reduce potential distress. 11 The same is true for handling and restraint procedures. Behavioural conditioning of the animals, such as adaptation to handling or structured positive reinforcement training may prepare the animals better for procedures and mitigate these effects,
Administration of substances and sampling procedures may have a negative impact on welfare, in the short or long-term dependent on the routes, volumes and the effects of the administered substances.
Surgical interventions are likely to cause some pain, even with good peri-operative care (including the use of analgesics).
How much suffering will these interventions cause?
Consideration needs to be given to all the individual elements, and how these will interact.
The nature, intensity and duration of each intervention will impact on the overall severity.
The frequency of interventions and recovery time between interventions also need to be considered.
Duration of the study is an important factor to consider and the period over which the animal may experience pain, suffering or distress. For example, in a safety evaluation/toxicology study, depending on the dose, an acute study may cause major discomfort as a consequence of drug administration but this would generally be of short duration. In contrast, an animal may be exposed to contaminated material (e.g. scrapie/BSE) as a juvenile with no initial adverse effects and, due to the very long incubation period, will remain in good health until the onset of clinical disease.
In chronic toxicology studies, animals may experience minor/moderate adverse effects over many months as a combination of daily dosing and the effects of the test substance.
For work in the wild, a careful scrutiny of the project authorisation is extremely relevant, as severity classification of only animals used and as described under project authorisation is to be reported. 7
When, in agreement with Article 10, exemptions are given to use wild animals captured from nature, it is then
It goes without saying that the appropriate welfare during capture and transport under the Directive must be ensured: the capture may only be carried out by competent person(s), using methods which do not cause avoidable pain, suffering, distress or lasting harm; animals must be transported under appropriate conditions using appropriate methods of containment; any animal found to be injured or in poor health shall be examined by a veterinarian or other competent person and actions shall be taken to minimise suffering. Special considerations shall be given and appropriate measures taken for the acclimatisation, quarantine, housing, husbandry, and care of animals taken from the wild and, as appropriate, equally provisions for setting them free at the end of procedures.
What interventions can be included to reduce the impact on the animals?
How can suffering be minimised? How are the principles of the 3Rs embedded in the procedure/project?
Plan to minimise disruption to accommodation, husbandry and care practices. Develop processes for acclimatisation, implement training programmes as appropriate. Consider dosing and sampling procedures to minimise impact on animals. For example, mini-pump administration may have reduced adverse welfare impact compared with multiple daily dosing, which may require stressful restraint and acclimatisation to handling. However, this needs to be balanced with the need for surgery and the relative size of the implant which may impact on locomotion and/or behaviour. Surgical interventions must be accompanied by effective peri-operative care, and appropriate pre-emptive and post-operative analgesia. During the planning of procedures, consider the development of welfare monitoring/scoring systems and identifying likely clinical effects and determining early end-points (consistent with scientific objectives).
Each element of a procedure should be challenged to ensure that the 3Rs have been properly addressed. These issues should be considered initially by the Project Applicant, and should be informed by discussions with the veterinarian and animal care staff, and the AWB before the details of the procedure are finalised and the severity assigned in the application.
Prospective discussions with all relevant personnel will ensure that the most refined procedure is developed.
Discussions should continue throughout the procedure to ensure that all opportunities are taken to further develop and adopt refinements as these become available, for example through new publications.
Retrospective assessment (RA) and assessment of ‘actual’ severity
As indicated earlier, there are requirements in the Directive for the assessment of actual severity experienced by each animal and, for certain projects, a requirement that an RA shall be performed (Article 39).
Assessment of actual severity is necessary for a number of reasons:
to provide information for the annual statistical returns on animal use; to enable consideration of requests for re-use of animals; to contribute to the RA of projects (where required).
RA, in addition to the consideration of actual severity experienced by the animals, also requires consideration of whether or not the objectives of the Project have been achieved, and whether further opportunities for implementation of the 3Rs have been identified. All projects using non-human primates and all those involving ‘severe’ procedures
RA may be required for other projects – these are determined by CA during project evaluation and the applicant informed where and when RA is required.
In order to be able to determine ‘actual’ severity there is a need to develop recording and assessment systems, tailored to each project which will capture all the necessary information in a format which will facilitate subsequent assessment and categorisation of the actual severity.
Development of a system for the monitoring and assessment of welfare
There are many publications that offer guidance on the assessment of welfare in animals undergoing scientific procedures (see references below). The Working Group refers readers in particular to three articles on the creation and use of follow-up and evaluation sheets, namely articles by Morton, Buchanan-Smith and the report by the BVAAWF/FRAME/RSPCA/UFAW Joint Working Group on Refinement.12–14 These articles not only provide background on this topic, they discuss the benefits of using such sheets for the experimental animals, the animal care staff, and the science of a project. Other useful references to consider are the Guidelines for the Assessment and Management of Pain in Rodents and Rabbits, the Rabbit Grimace Scale – a new method for pain assessment in rabbits and the 1994 FELASA publication giving guidelines on pain and distress that offers general guidance on clinical signs in rodents and lagomorphs, equating to a severity category.15–17
An ideal assessment system would include simple, objective measurements which could be applied consistently and used to detect the onset, and monitor the development of pain, suffering and distress in animals undergoing scientific procedures. Unfortunately, such a system is not available, nor is likely ever to be so, due to the wide variation in behaviours and behavioural responses among different species, strains and individual animals and the specifics of procedures.
The use of tailored assessment systems specific to the project, using trained and experienced personnel can contribute to significant refinements in animal models. Thus, it is important to list possible and/or observed behavioural or clinical responses, which moreover should be assessed and quantified or scored wherever possible, and may allow for identification of humane end-points. For example, in some infection studies body-temperature monitoring has been successfully used to identify early suitable end-points in advance of clinical evidence of disease, while still allowing the scientific objectives of the study to be met. 18
The following points should be considered in the development of a monitoring and assessment system:
Targeted at an individual animal and not at a group of experimental animals, although this may be challenging where large groups are involved (e.g. in some fish studies). Level of severity that is experienced by each animal needs to be reported using the categories of mild, moderate, or severe. Consideration should be given to the administrative burden in the design of the recording system System should, where possible, use objective measures to assess the level of pain, suffering or distress experienced by the animal during the procedure. System should require a definition and description of humane end-points. The monitoring must capture (a) any welfare-related issues, both expected and unexpected, that occurs during the course of the project, (b) any refinement actions that were taken during the course of a project. Assessment criteria should be included to facilitate the severity classification. Many of the published systems advocate some form of numerical scoring system, and rank clinical signs with severity allocation. Evidently, expertise and professional judgment will better allow for objective scoring. Standardised recording is essential. Although it is acknowledged that all scoring systems will contain subjective criteria to some degree, the information recorded should be specific for the model and species used.
Development of a suitable recording system
Develop an animal welfare assessment sheet tailored to the research project through discussions with researchers, care staff and veterinarian (or suitably qualified expert where more appropriate). Where applicable, score the signs of discomfort on a convenient scale from normal (score = 0) to the highest level of severity. Use objective scoring where possible. Identify intervention criteria – for example state signs which require veterinary check/intervention Define the limit of acceptable or permissible severity (e.g. a maximum score of clinical signs/behaviours for the procedures that should not be exceeded. This score can then be used as the score for adopting a humane end-point.) Include consideration of the assessment of cumulative suffering and criteria for re-use where applicable An electronic format may facilitate data entry, allowing the details to be modified at any time in order to permit the recording of unexpected events and any new events when they occur during the project and also allows easy sharing of information with all those involved (technicians, researchers, veterinarians, AWB, etc.). The assessment sheet should be simple and easy-to-use for experienced and inexperienced or novice observers, based on and using agreed terminology (e.g. FELASA Glossary of clinical signs).
19
The assessment sheet should be structured in such a way that the results of different moments of observing the experimental animals, such as on handling, close up, or from a distance, can be recorded. The assessment sheet should be structured such that it can be easily modified for changing situations, type of project, and animal species. The assessment sheet should, when required, allow the recording of the time when each procedure, technique was performed, or refinement introduced during the project. The assessment sheet should be useful for the entire duration of a project. Records of such assessments will be useful for subsequent review of the project and tailoring improvements for future studies.
Assessment of actual severity
An animal’s overall or cumulative suffering can be estimated from the nature and number of adverse and unexpected effects that appeared during the course of a procedure. The level of severity experienced by each animal during the course of a procedure is influenced by several factors, each of which should be incorporated.
The non-exhaustive list below gives an indication of the factors which can influence the amount and level of suffering that an experimental animal may experience during a scientific procedure, and which need to be taken into account when determining cumulative suffering of an experimental animal:
the duration of the project/procedure; the duration of any adverse effect the number of procedures that were carried out on the animal; the frequency of performing the procedures; information on whether the animal used in this project is being re-used an assessment of the animal’s clinical condition and physical wellbeing at the end of the procedure, which should include determination of those factors that influence body weight and body condition; an assessment of the impact on the animal’s behaviour or psychological wellbeing, for example, the incidence of abnormal, stereotypic or aggressive behaviours.
The following factors could also be given consideration in the assessment of actual severity:
how the animal was conditioned (e.g. adaptation, training); the number of (surgical) interventions; the routes, volumes and frequencies of compound and drug administration; the physical and chemical characteristics of the administered compound or solution, for example, whether the repeated injections or the injections of acidic or basic substances induced local irritation and necrosis; the routes, frequency and volume removed during blood samplings; the method and frequency of restraint; changes in social structure/separation and single housing of social animals.
Although each element has the potential to impact on severity, the actual severity experienced will largely be determined by the effectiveness of the actions taken to reduce the negative impacts of the procedure – for example, the use of analgesics will reduce post-operative pain.
Determination of actual severity requires a review of the application and consequences of the applied procedures and the effectiveness of actions taken to minimise suffering. The actual severity can only be determined following a review of all the effects on the animal throughout the procedure – this necessitates the maintenance and consideration of focussed clinical records.
The actual severity to be reported for each individual animal should be based on the highest level of severity experienced during the course of the procedure and not based on the severity at the end of the procedure.
Assigning severity to an animal found dead during study
Despite the best efforts to monitor animals closely, it is possible that an animal might be found dead as a consequence of either the experimental procedure or other unrelated causes. Some guidance on assignment of severity in such cases is given in the Commission working document on a severity assessment framework and in the 2016 Discussion paper.6,7 These state that: ‘For the purposes of statistical reporting,
All deaths of animals should be carefully reviewed among those involved (e.g. scientist/care staff/veterinary surgeon) as soon as possible to ensure that all relevant information is available to determine whether or not the death was procedure related and to determine an appropriate level for reporting purposes. Whatever the cause, measures should be taken to avoid recurrence.
When the actual severity experienced exceeds that predicted prospectively for the procedure, there may be a need to notify authorities and/or revise project authorisations.
Further guidance on the assignation of actual severity where animals are found dead can be found in the EU discussion paper of January 2016, which provides an illustrative decision tree to assist determination in assigning the severity of death for the purposes of statistical reporting (reproduced below).
7
Is the death unrelated or related to the procedure the animal was undergoing?
1.1. Unrelated
Examples of unrelated deaths:
deficiencies in equipment or environmental controls such as cage flooding, heating/ventilation malfunction; inappropriate husbandry or care practices such as failure to provide adequate diet (e.g. inappropriately balanced) or diet contaminated (e.g. poor storage); aggression between animals in a group housing; unrelated disease and infections; Ageing animals: deaths in animals on long-term studies should be evaluated to In the case of GA breeding of an established line, when the The actual severity for the animal should reflect the highest level of severity experienced during the course of the procedure by the animal (excluding the level of severity related to the death). 1.2. Related: proceed to question 2. 2. 2.1. • animal failing to fully recover consciousness in post-operative period, but under appropriate analgesic regime throughout; • no clinical abnormalities recorded throughout the procedure, nor anticipated, but found dead a few hours after a clinical examination. The actual reported severity should reflect the severity as the result of the assumed events leading to death. The actual reported severity should reflect the severity as the result of the assumed events leading to death. 2.2. The actual severity should be reported as ‘severe’. The actual severity for the animal should reflect the highest level of severity experienced during the course of the procedure by the animal (excluding the level of severity related to the death). The actual reported severity should reflect the severity as the result of the assumed events leading to death. The actual severity should be reported as ‘severe’.
Re-use of animals and cumulative suffering
Re-use of animals in further procedures is permissible under the Directive 2010/63/EU, but is dependent on a number of factors including the actual severity of the previous procedure, a demonstration that the animal’s general state of health and well-being has been fully restored and that re-use is in accordance with veterinary advice, taking into account the lifetime experience of the animal. 20
Assessing the severity that an individual scientific procedure will cause to an animal can be difficult when animals undergo several multi-step procedures over prolonged periods, especially when the nature of the procedures means that the animals may also be subjected to alterations in normal housing and care practices (e.g. periods of single housing).
However, such an assessment is necessary to allow re-use, and this needs to take account of the animal’s lifetime experience. This introduces a further area for consideration as now not only does the direct pain, suffering or distress caused by the various steps in the procedure need to be taken into account, but also some consideration is needed of any contingent suffering due to the animal’s husbandry and care environment throughout its lifetime.
Lifetime or cumulative suffering can be considered as the combination of direct suffering (the application of scientific procedures), any clinical conditions from which the animal has suffered (which may or may not be due to the procedure being carried out, e.g. inter-current disease or surgical wound) and contingent suffering (housing, husbandry, transport etc.); the duration of these events must be taken into account.
The key issues which need to be taken into account when considering lifetime experience are:
the duration of exposure to the pain suffering distress or lasting harm – longer duration is more likely to cause higher severity; the nature and intensity of the effects on the animals; the interval between procedures – the shorter the interval (usually) the less opportunity the animal has to return to normal; the nature of interventions and actions that will be taken to relieve the suffering; consideration of any contingent suffering.
Illustrative examples of severity classification and reporting - Appendix 1
The attached worked examples (Appendix 1) were current during the evolution of this report and were real examples of how severity classification and reporting can be approached. They highlight the welfare and scientific issues to be considered, suggesting improvements that can be made through critical review of a study design, and provide illustrative realistic severity classifications. The examples also include illustrations of recording systems that can be used during the course of studies to monitor and assess actual severity and contribute elements to retrospective assessment of a project.
It is acknowledged that there may be further 3R opportunities that have evolved since these were developed, and therefore these illustrations are not intended to be used unaltered by research workers. Each project will be different in particular with regard to scientific objectives that can influence significantly the overall severity of a procedure. The intention however is to explain and promote this stepwise approach to severity assessment. If applied as intended, the desired outcomes of improved science and welfare and consistent assessment and reporting of actual severity should be achieved.
The WG has chosen animal models commonly used in the scientific community and has for each of them addressed the previously stated four questions asked namely: What is being done to the animals? What effect will this have on the animals? How much suffering may it cause? What interventions can be included to reduce the impact on the animals?
Models included in this report
Control of infection: assessment of protection of vaccine candidates in a murine model of tuberculosis and screening of novel drug candidates Neuropathic pain: spinal nerve ligation in the rat Stroke: efficacy of a novel therapeutic agent on intraluminal thread middle cerebral artery occlusion (MCAO) in the marmoset Cardiovascular evaluation of novel therapeutics: telemetered dog model Atrial fibrillation: evaluation of novel antiarrhythmic substances in the rabbit Ecotoxicology: determination of bioaccumulation using the fish flow through test Regulatory toxicology: assessment of acute oral toxicity in the rat Pharmacokinetics: determination of the pharmacokinetics after a single administration of a test substance in the dog
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the publication of this article: Publication of our work was supported by a bursary from Laboratory Animals Limited.
Acknowledgements
The authors would like to thank Laszio Dezsi for technical assistance with the spinal nerve ligation model and Pascale van Loo for technical assistance with the pharmacokinetic model.
References
Appendix 1: Illustrative examples of severity classification and reporting
Control of infection – murine models of TB
General context
In 2010, 8.8 million cases of active pulmonary disease were identified, with around 1.2–1.5 million people dying of this disease in that year, making it the second largest cause of infection-related deaths worldwide, after HIV/AIDS. 1 Although there are some effective drug regimens available for treating human TB, there are presently some hard-to-tackle challenges in the fight against this infection, that include the rise in co-infection with HIV, the emergence of multidrug-resistant strains of Mycobacterium tuberculosis, compliance problems with current long-term drug regimens and the need for new vaccines to replace BCG, which efficacy has been reported to vary between 0 and 80%.1–4 This makes the use of murine models of TB in pre-clinical as important as ever.5,6
Experimental infection of mice with M. tuberculosis has been used to model human TB since the early works of Robert Koch, and these models have since then been of pivotal importance for the understanding of host-pathogen interaction and for testing therapeutic and preventive approaches to this disease.5,7–10
There are marked differences in susceptibility to TB infection between mouse strains. However, and contrary to what happens in most humans, no mouse strain is capable of controlling disease to a truly latent state, and all animals eventually succumb to the infection as a result of progressive disease, if left untreated. 11 In all strains experimental infection is quickly followed by an accentuated and continuous growth of bacterial numbers in the lungs. The more resistant strains (e.g. the C57BL/6) are capable of mounting a specific immune response after this primary response, being thus able to control the disease to a chronic stage from 3–4 weeks post-infection. During this stage bacillary numbers in the lungs remain high but relatively stable for several months and up to more than a year (although lung pathology ensues) and animals are seemingly asymptomatic. Eventually, disease recrudesces, progressing in severity until death,1,12 if not averted by humane end-points. 13 As for the more susceptible strains, these either fail to inhibit bacillary growth in the lungs after primary infection or cannot maintain it, resulting in rapidly progressive and overtly symptomatic disease, which culminates in early death.14,15 Aside mouse strain, other important parameters affect resistance to infection, such as the inoculum size (in CFUs) and the chosen route of infection.11,16,17 Depending on these various parameters, median survival times of M. tuberculosis-infected mice may vary between less than 20 days to more than 300 days.11,18
Two different procedures are described below, one for vaccine testing and the other for screening of novel drug candidates.
Illustrative procedure (1) – assessment of protection of vaccine candidates in a murine model of TB infection
Study design
Consideration of specific refinements and humane end-points

Initial prospective assessment
While bacillary burden and immunological parameters are to be used as indicators of vaccine protection (see interim evaluation), measurement of survival provides important data and is commonly used in this sort of study. Objective criteria, as assessed by clinical scoring, will however be used to implement humane end-points to prevent animals of reaching advanced stages of disease. Nevertheless there is the risk of at least some animals unpredictably reaching significant levels of suffering and distress as a result of experimental infection (particularly vehicle controls), as well as from accidental nerve damage or exacerbated immune response, hence swelling from antigen inoculation (cf. 3 i.m. injections in the thigh of mice).20,21 A prospective severity category of
Could the severity category be
Not if the proposed refinement measures are properly applied. The use of the scoring sheets in particular may prove valuable for identifying early signs of active disease and thus allow identifying early humane end-points, preventing animals from significant suffering.
Could the severity category be
Depending on the time point of euthanasia, animals may not yet show signs of onset of disease therefore classification would be MILD.
For survival studies, in cases where the vaccine control has been effective and refinement opportunities maximised, there is the possibility that those animals may only exhibit MILD clinical signs. However, as in mice, even BCG vaccination only reduces bacillary burden (see introduction) and thus eventually all lose control of the infection a severity classification of MILD is unlikely.
Clinical observation/scoring system
Results and assessment of actual severity
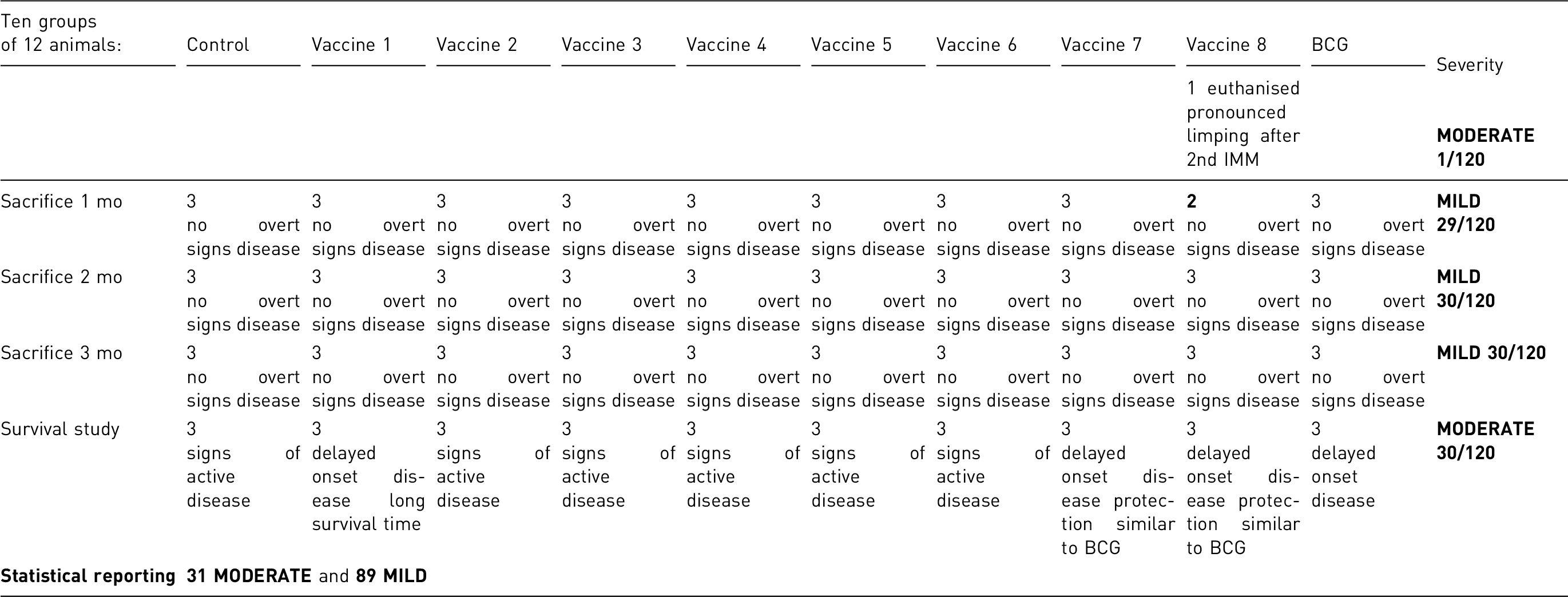
Apart from one animal that had to be euthanised and removed from the study for showing a pronounced limping gait in one hind leg after the second immunisation, all animals recovered from the immunisation scheme with no unexpected complications. No complications were associated with aerosol infection.
All mice euthanised at the predefined time-points (three per group, three time-points) showed no overt signs of disease.
Of the eight vaccinated groups, five of the vaccinated groups showed lower bacillary burden in all organs analysed, a delayed onset of disease and a significantly longer survival time than non-vaccinated groups, two vaccinated groups showed levels of protection similar to the BCG group and in the one remaining group pathology resembled that of controls, which were the first to show signs of recrudescent disease. Despite differences in time-of-onset, rate of progression and survival, all animals in the survival study eventually showed signs of recrudescent disease and were timely euthanised according to the predefined clinical score. No unexpected animal loss (e.g. spontaneous death) was registered.
89 animals (euthanised at the predefined time-points except the one limping) were considered to have experienced MILD severity.
Example clinical observation/scoring system

A total score of 10 signals the humane end-point, at which the animals will be euthanised.
Illustrative procedure (2) – screening of novel drug candidates in a murine model of TB infection
Study design
The aim is to find drugs that can match current effective antibiotics, and allow all animals to survive up to six months without relapse, post-infection. For this period, we have observed a survival of 20% in untreated controls. To detect such a survival difference, with a 90% power and an alpha = 0.05, only six animals are needed (Fisher’s exact test). However, to accommodate unexpected losses, eight animals will be used per group. 13 groups of n = 8 TB-susceptible C3H/HeJ female mice will be used to compare the efficacy of four novel drug candidates against M. tuberculosis infection with existing licensed products. All mice will be infected via the intratracheal route (3 logs of CFU in 100 μl PBS, a dose deemed appropriate during preliminary testing) by an incision in the trachea. Four weeks post infection, four groups of mice will be administered a low dose of their assigned test compound daily five days a week by oral gavage, for two weeks. Four groups will be given high doses of the test compound. One group of control mice will be administered vehicle only, and four other groups will be given either one of two drugs currently used as gold-standards (GS-A, GS-B; positive controls), in two doses for each drug. Survival will be recorded for all groups of mice. A clinical score will be used to define humane end-points. Mice will be housed in solid floored cages with litter and nesting material and cardboard tubes. Animals will be provided food and water ad libitum. All surviving animals will be euthanised by anaesthetic overdose with pentobarbital sodium at the end of the experiment (14 days after dosing).
Consideration of specific refinements and humane end-points

Initial prospective assessment
The use of intratracheal instillation, when compared with other routes of infection, raises additional welfare issues since it requires general anaesthesia and, aside recovery-associated distress, complications may arise before full wound cicatrisation.23 However, it allows for an accurate, standardised and successful inoculation. An optimised surgical procedure will aim to avoid post-surgical complications and reduce variability.
While a measure of survival is required, death will be replaced by humane end-points based on objective and easily measurable clinical parameters. Nevertheless, as C3H mice cannot establish a long-lasting control of M. tuberculosis infection, there is the possibility of at least some animals reaching advanced stages of disease at the time of treatment onset (particularly the vehicle controls), or as a result of low experimental treatment efficacy.
A prospective severity classification of
Could the severity classification be
Where disease progress is interrupted by drug treatment in gold-standard controls, and possibly also in test-compound-treated groups, these animals, may retrospectively be found to not have exceeded the MODERATE category.
Could the severity classification be
No – surgical intervention under general anaesthesia is by definition considered MODERATE.
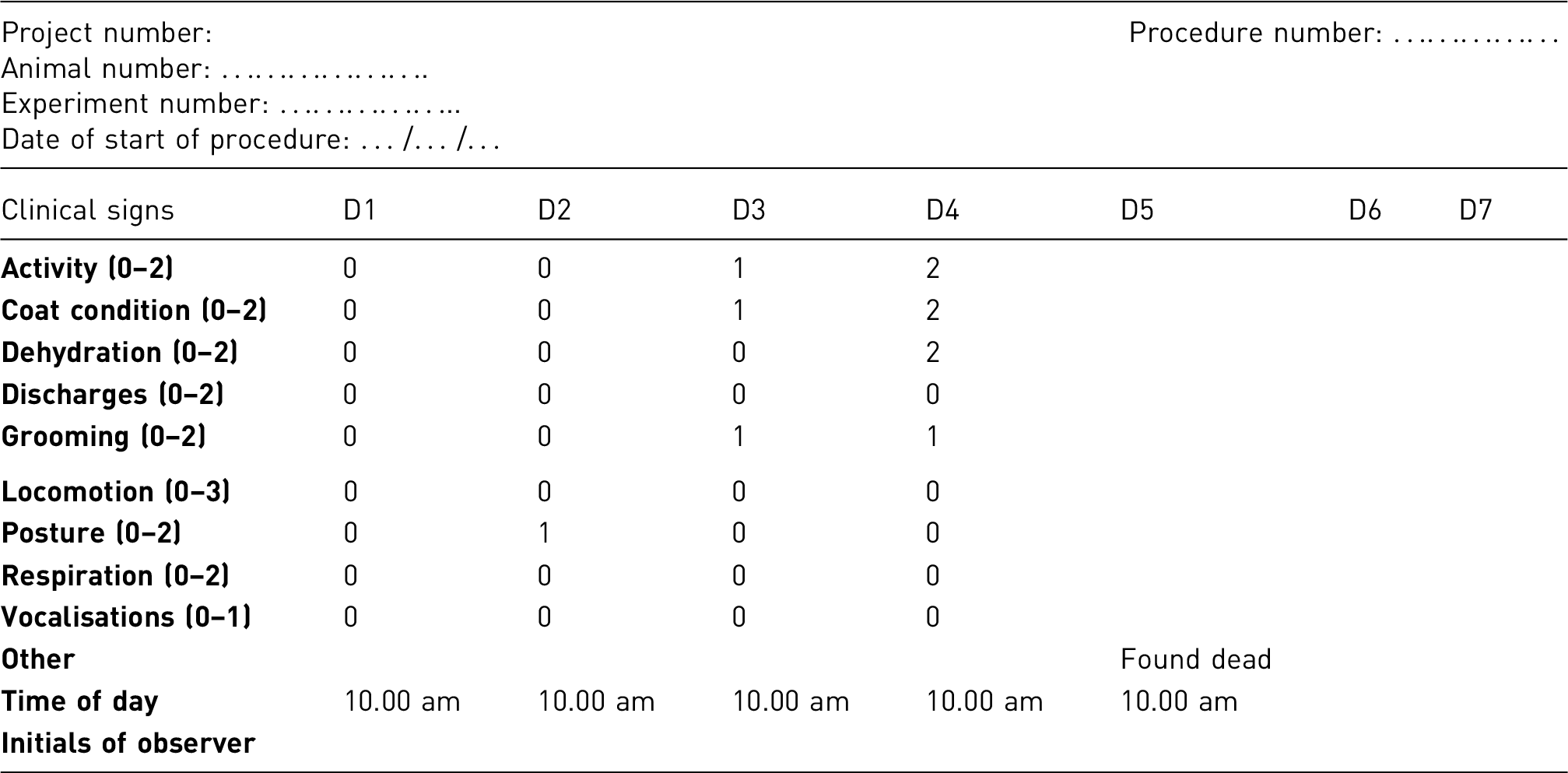
Clinical observation/scoring system
Animals are very carefully monitored; analgesia and local supportive therapy are provided as necessary.
The combined clinical observation and scoring system used to help monitor the clinical condition of the animals throughout the procedure is the same as that used in the preceding example.
Results and assessment of actual severity
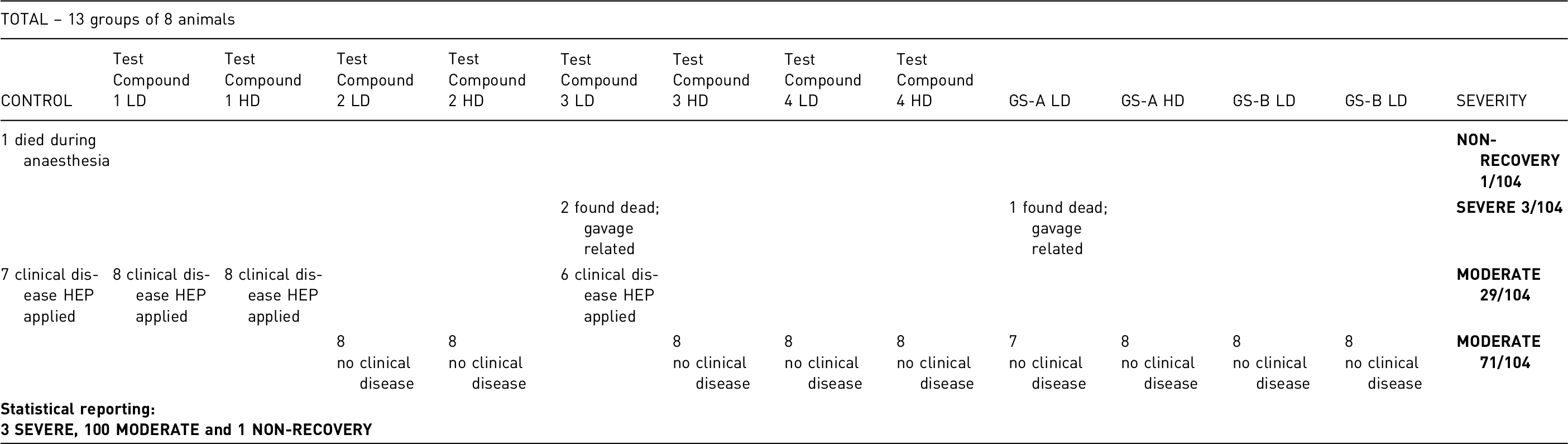
One animal died under anaesthesia. All other animals survived and recovered from intratracheal instillation without any complications from this procedural step. Gold-standard controls showed no signs of disease at the end of the treatment. The proposed pilot study showed animals to be more compliant with oral gavaging when sugar-coated gavage needles were used. Nevertheless, three animals were found dead at the end of the drug treatment (one from gold-standard controls, two from Test Compound 3 LD), with post-mortem analysis showing gavage-related injuries as the most likely cause.
Two groups of mice treated with test compound 1(low and high dose) showed results comparable to the vehicle controls, along with one group with low dose of test compound 3. All of these animals reached symptomatic stages of disease, but spontaneous death was prevented by close monitoring for humane end-points. Test compounds 2 and 4 prevented disease development in both low and high- dose groups, and test compound 3 only in high-dose treated animals.
1 animal died under anaesthesia during initial immunisation (from negative control group):
71 (31 low and high-dose gold-standard drug controls + 32 low and high dose test-drug mice and 8 high-dose test-drug mice): although disease development was prevented and no clinical signs developed, due to the surgery it is
29 animals (7 negative controls + 16 low and high dose test-drug mice and 6 low-dose test drug mice):
3 gavage related incidents (1 animal from gold-standard group, 2 from the same low-dose treated group):
References
Neuropathic pain – spinal nerve ligation
General context
Neuropathic pain (NP) refers to a group of pain syndromes (e.g. spontaneous burning pain, allodynia, hyperalgesia, aftersensation, paraesthesias, etc.) that result from an initial nerve injury that causes an increased responsiveness and pathological signal transmission in the pain pathways of the nervous system. The estimated prevalence of NP is about 1–1.5% in the population. The largest group of patients are those with diabetic peripheral neuropathy (representing about 45–70% of diabetic patients) while post therapeutic neuralgia is the second most common cause of NP. AIDS and cancer/chemotherapy may also predispose to NP. Traumatic nerve injury may lead to a pain syndrome termed causalgia or complex regional pain syndrome II (CRPS II) which is considered as the most severe NP conditions. NP is resistant to conventional pain medications, and as such represents a major therapeutic challenge. In spite of recent improvements in therapy by the introduction of certain novel drugs, there is still a huge unmet medical need for new medications with higher efficacy, more rapid onset of action and better side effect profile.
A typical and hardly tolerable NP symptom is allodynia, when a mechanical or thermal stimulus (e.g. skin contact by clothes) which does not normally provoke pain becomes painful. Drugs with anti-allodynic effects may substantially alleviate the suffering of neuropathic patients.
Experimental nerve injury in animals can be induced by several methods which mimic different NP aetiologies. 1 Animals with streptozotocin-induced diabetes also develop painful peripheral neuropathy, while toxic polyneuropathy can be modelled by cytotoxic drug treatment, e.g. vincristine or cisplatin. Various models based on surgical injury to a major nerve trunk (partial ligation or transection) have also been developed which are more or less direct translations of human CRPS II. The model under analysis uses spinal nerve lesion to induce mechanical allodynia for testing the effects of analgesic compounds. 2 A unilateral ligation of the fifth lumbar (L5) spinal nerve is performed, and then animals are allowed to recover for 2 weeks. The advantage of the spinal nerve lesion model over other nerve injury models such as the chronic constriction or the partial ligation of the sciatic nerve is that the degree of injury is more uniform and therefore the development of allodynia is more consistent. Moreover, the motor deficit and the foot deformities after the selective L5 lesion are less severe. The mechano-nociceptive threshold of rats are determined by dynamic plantar aesthesiometer and/or von Frey filaments before and after surgery. 3 Thermal allodynia can also be measured with radiant heat paw stimulator. Test compounds are usually administered under a repeated dose regimen starting after the recovery period when allodynia has already developed.
Illustrative procedure
Study design
In this example, 30 male Sprague-Dawley rats will undergo unilateral spinal nerve ligation. After the operation they will be allowed to recover for 2 weeks, during which the condition of the wound, the affected limb and the general health status including body weight measurements and observation of home cage behaviour will be monitored daily. On the 14th postoperative day mechano-nociceptive thresholds will be measured to confirm the development of mechanical allodynia. Only animals having a minimum of 20% decrease of the pre-operation threshold measured by the dynamic plantar aesthesiometer and a von Frey threshold <5.4 g are included in the treatment groups. Allodynic rats are randomised to form three treatment groups to be treated intraperitoneally (i.p.) with test compound A at a low dose, at a high dose and a vehicle control, respectively. An 8-day-long repeated dose regimen is utilised with daily treatments and the anti-allodynic effect is determined on Day 1, Day 4 and Day 8 of treatment at 30 and 60 min after the injection. At the end of the study animals are euthanised. In this study, no analgesia will be provided as this would interfere with the study results.
Consideration of specific refinements and humane end-points

Initial prospective assessment
Interventions involved in this model individually do not exceed moderate severity. If the surgery is carried out with proper skills and, consequently, no complications occur, this part of the procedure is within the moderate category. Following surgery, careful monitoring allied to clear end-points will ensure no animals exceed moderate severity.
A prospective severity classification of MODERATE is therefore appropriate.
Could the severity classification be MILD?
No. As the prolonged pain resulting from these interventions renders the model moderate, and assessment of the pain itself is the objective of the study, it is not possible to conduct this procedure within a MILD classification.
Clinical observation/scoring system
Animals were carefully monitored from surgery until the end of the procedure. Two nociceptive assays were applied to measure latency of hind feet withdrawal: (1) von Frey hairs of different stiffness were used to determine the one that evoked a hind paw withdrawal; (2) dynamic plantar aesthesiometer. During the assays no additional agent was used.
An example of an observation sheet and a sample score sheet to help monitor the clinical condition of animals throughout the procedure are included at the end of this example.
Results and assessment of actual severity
All animals, except one in the vehicle treated group, recovered from surgery with no unexpected complications, due to the intensive peri-operative support provided.
Nociceptive assays indicated that mild or moderate pain was experienced.
Vehicle group
1/10 animals did not recover from surgery. NON-RECOVERY
1/10 animals showed signs of automutilation and was euthanised. MODERATE severity.
1/10 animals reached a humane end-point and was euthanised. MODERATE severity.
7/10 animals showed a poor performance in the nociceptive assays and the behavioural tests compared to treated animals. However they did not show any other clinical effects and maintained body weight. Clinical score was similar to treated animals after the surgery. These animals developed moderate neurological-locomotor deficit, and showed a gradual reduction in clinical score over time, possible resulting from their ability to compensate and adapt to long term neurologic deficits. MODERATE severity.
Treatment groups
10/10 animals treated at lower doses showed mild improvement in motor function, together with an improvement in clinical scoring. The agent had anti-allodynic effect, compared to vehicle. No specific side-effect was reported. MODERATE severity.
10/10 animals treated at higher doses showed significant improvement in motor function, together with an improvement in clinical scoring. The agent had clear anti-allodynic effect, compared to vehicle. No specific side-effect was reported. MODERATE severity.
Although animals in the treated groups experienced less pain, due to the surgery and prolonged allodynia the severity category for all animals was considered to be moderate.
Example clinical observation/scoring system

HEP: humane end-point
Score 0–5 plus surgery = MODERATE
Either
Note: that as surgical complications are generally noted in the immediate post-op recovery period, close monitoring and expert, empathetic judgement are essential during the first 24 h to ensure that adverse effects are identified and actions taken to address these. Animals are humanely killed if their suffering exceeds of the moderate category.
1 – Review frequency of monitoring.
4 – Provide appropriate supplementary care, e.g. mash and additional fluids
Dehydration/diarrhoea: Ringer Lactate or regular serum
Abdominal dilation (ascites): draining for pressure reduction
Weight loss: soft food
5 – Review progress with vet
Either
References
Stroke – efficacy of a novel therapeutic agent on intraluminal thread MCAO model in the marmoset
General context
Stroke is defined as loss or alteration of normal body function that results from an insufficient supply of blood to part of the brain. Despite better understanding of the pathophysiology of vascular brain injury, an effective treatment for stroke remains an important unmet medical need, and research is on-going to find appropriate preventive and therapeutic measures.
Three different types of stroke can be seen in human patients: ischaemic, intra-cerebral haemorrhage and subarachnoid haemorrhage, but most of the animal models currently available are based on the ischaemic type. Stroke models, by their very nature, represent a challenge from the perspective of animal welfare. Good interactions and communication between all individuals involved in the scientific procedures, (veterinarians, investigators, animal technologists and care staff), are critical to ensure that there is adequate balance between achieving a valid model in this research area and minimising animal suffering.
Stroke is routinely induced in rodents and non-human primates by temporarily or permanently occluding the middle cerebral artery (MCAO model).This ‘MCAO’ model aims to reproduce experimentally the focal cerebral ischemia that occurs in stroke, and it has been extensively used to study the mechanisms of injury, to identify potential targets and to test putative neuroprotective agents.
In a standard study design, the animals are trained to perform certain behavioural tests prior to the MCAO procedure. During the therapeutic time window, established according to the mechanism of drug action and objective of the study, animals are given the test compound. The outcome analysis should include information on infarct size, mortality rate, frequency of complications (e.g. subarachnoid haemorrhage), together with functional and neurological evaluation to monitor progress. Serial magnetic resonance imaging (MRI) has proven to be a powerful tool to gain information on variation of infarct size over time, but can also provide additional information on blood flow or metabolic state. Histological, biochemical and molecular end-points can also be included.
There are various behavioural tests that may be applied to stroke models. The simplest tests include neurological scoring systems, which assess global neurological status, and limb placing tests, used to measure motor reflexes. These are generally used to assess animals in the acute post-stroke phase. In long-term studies, more complex tests may be used to assess sensory and motor functions (e.g. bilateral sticky label test, beam walking, rotarod or staircase) and cognitive functions such as memory (e.g. passive avoidance tests, or evaluations of learning strategies).
It is good practice to perform a group of behavioural tests, including at least one for each phase (acute and long-term), so as to gather comprehensive information on the impact on sensory, motor and cognitive functions. These tests have to be carefully chosen to capture any effects of the putative therapeutic strategies. Detailed descriptions of each of these behavioural tests, including training schedules, are not included here, but for a comprehensive review and discussion of their use see Schaar et al. 1
Many recent recommendations for preclinical investigations designed to develop stroke therapies recommend the use of higher order species such as non-human primates in addition to rodent models.2–5 The common marmoset (Callithrix jacchus) may be considered the species of choice to study the pathophysiology and the treatment of cerebral ischemia. Compared with rodents, this primate is closer to humans in term of cerebrovascular system, brain metabolism, grey-to-white matter ratio and has a richer behavioural repertoire. In addition, compared to old world monkeys, they are easier to handle, which is advantageous for behavioural testing and postoperative care management. The location and anatomy of the MCA in the marmoset have historically restricted the approaches used in rats and thus a more invasive surgical model was developed that included the turning of a large bone flap to access the brain and the MCA. Nevertheless, more recently the intraluminal thread approach has also been described in the marmoset. 6 The intravascular approach presents a number of advantages compared to previously used methods, and should be considered a clear refinement. In particular the absence of craniotomy and the comparative non-invasiveness results in fewer adverse effects (e.g. severe disability/mortality) encountered in the postoperative period.
As in rodents, the first 48 h post-surgery are critical. The animals will have difficulties caring for themselves, and typical impairments can include left arm hemiparesis, abnormal grasp reflex, left-sided neglect, nystagmus and rotation of eye. 7 Generally, after 3–7 days the animals are capable of self-care and can return to their home cage. Gradually (around 2 weeks post-surgery), they will recover the majority of motor abilities and will be able to freely jump and climb around their cages.
Illustrative procedure
Study design
In this example, on efficacy of a new compound, six male and six female laboratory-bred marmosets (Callithrix jacchus) will undergo 3 h-transient MCAO using the intraluminal filament technique under general anaesthesia. Before surgery, marmosets will be trained and tested on a number of neurological tests, which assessed general neurological function, motor ability, and spatial awareness. Immediately after tMCAO, marmosets will receive a bolus of saline (n = 6) or test compound A (n = 6), and osmotic mini-pumps will be implanted subcutaneously, providing 48-h saline or drug infusion. Sensory-motor deficits will be assessed weekly up to 45 days after MCAO, and MRI scans performed under general anaesthesia, at 1 h, D8 and D45. Animals will be killed 46 days after MCAO.
Consideration of specific refinements and humane end-points

Initial prospective assessment
This model is considered SEVERE because of the surgical procedure involved and the deleterious effects of the MCA occlusion on the welfare of the animal, especially during the first week. Nevertheless, intensive post-operative care in the first 48 h up to 7 days, and close monitoring of the subsequent phase can greatly contribute to reduce negative impact on animal welfare. From the experimental point of view, attention to refinement and standardisation of the individual steps in the procedure can lead to reduced incidence of complications and variability, and consequently better quality of the data obtained and therefore a reduction of the number of animals required.
A clinical score sheet should be agreed upon by the researcher, veterinarian and animal technologists to set up criteria for monitoring and euthanasia; it will need to include the neurological score, together with other clinical criteria such as body weight, ability to care for themselves or reaction to stimulus.
Could the severity classification be MODERATE?
Experience/training of personnel involved, veterinary supervision, and intensive care in the early post MCAO period together with agreed end points can significantly reduce the incidence of negative effects experienced by the animals.
Clinical observations/scoring system
Animals are very carefully monitored in the post-operative period. Analgesia and local supportive therapy are provided.
An example of a combined neurological/clinical scoring system which is used to help monitor the clinical condition of the animals throughout the procedure is included at the end.
Results and assessment of severity
All animals recovered from surgery with no unexpected complications. Clinical scoring in all animals was similar in the first 48 h after MCAO, and all of them received intensive peri-operative support. No significant ipsilateral deficit was observed after induction of ischemia in neither control nor treated animals. A partial recovering of contralateral neurological deficit was observed in all animals.
‐ All six vehicle-treated animals developed moderate contralateral neurological deficit, together with a poor performance in the behavioural tests compared to treated animals. Clinical score was nevertheless improving over time possible resulting from their ability to compensate and adapt to long term neurologic deficits Assessment: SEVERE ‐ 2/6 treated animals developed moderate contralateral neurological deficit, together with a poor performance in the behavioural tests compared to treated animals. Clinical score was nevertheless improving over time, possible resulting from their ability to compensate and adapt to long term neurologic deficits Assessment: SEVERE ‐ 4/6 treated animals showed a significant improvement in neurological scoring after 48 h post-MCAO, together with an improvement in clinical scoring. Assessment: MODERATE
In conclusion, 8 animals were considered as SEVERE and 4 animals were considered as MODERATE
Example clinical observation/scoring system
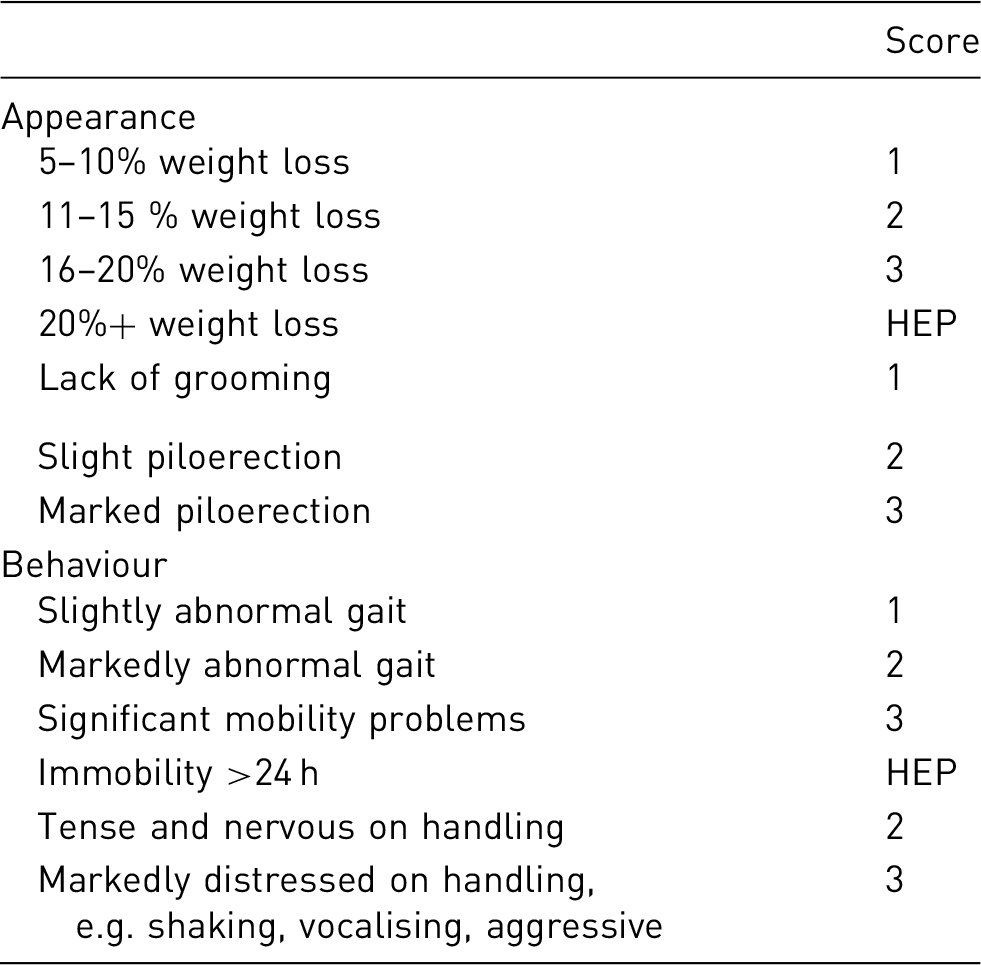
HEP: humane end-point
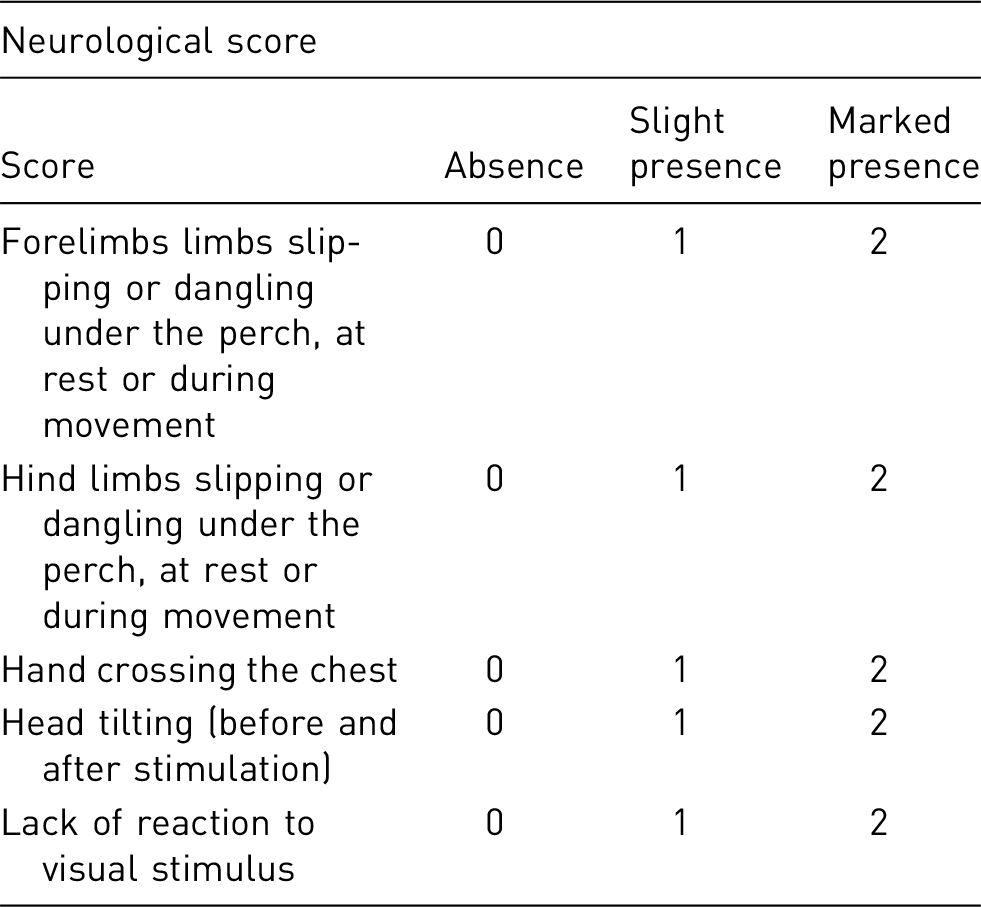
Each limb will be assessed and scored separately; severe contralateral neurological deficit together with poor clinical conditions are expected during the first week post-MCAO while ipsilateral neurological damage should be very limited/absent.
The neurological score for each limb will be added to the clinical observation scoring to obtain a cumulative score.
It is well known that immediately after MCAO, a high cumulative scoring is to be expected with a progressive improvement over the first week post- surgery. During this
≥ 22 – HEPtpb 1pc
1–10 – monitor regularly, evaluate together with the clinical score
10–20 – monitor frequently, provide care if not able to care for itself, evaluate clinical scoring and team to review all experimental data available (i.e. MRI, behavioural tests) to rule out unexpected complications, such as brain haemorrhage, oedema, etc.
≥ 20 – HEP
Example of an Individual observation sheet (Days 0–4)
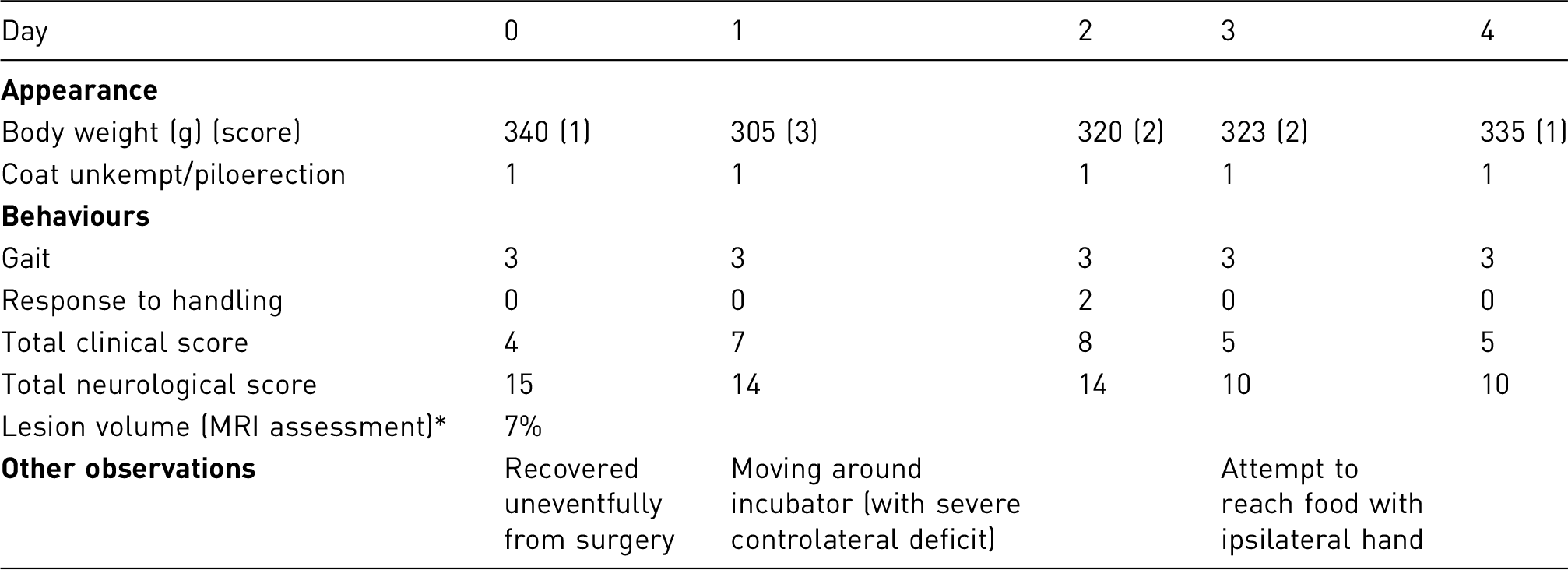
‘Lesion volume’ (assessed using MRI) is included for the investigator to fill in at the end of the study. This data can then be correlated with clinical and behavioural observations to enable further refinement of monitoring, animal care and procedures.
References
Cardiovascular evaluation of novel therapeutics – telemetered dog model
General context
Any medical drug to be marketed will need to prove efficacy but also safety. Therefore, even compounds intended for use in very different areas (e.g. neuropharmacology) will have to be evaluated for their potential cardiovascular effects.
Cardiovascular effects of compounds can be tested in a variety of ways: in invasive terminal procedures on anesthetised animals, in animals which are momentarily restrained and externally equipped with monitoring devices or in freely moving animals previously implanted with monitoring devices (telemetry systems). Over the past decades, telemetry systems have been increasingly applied in drug research and development for measurement of physiological and bioelectrical variables (e.g. blood pressure, heart rate, ECG). 1 The absence of tethering, handling and restraint during measurement provides a unique opportunity to study laboratory animals without additional stress or physiological disturbance (anaesthesia) during a longer period of time. In between measurements, animals can moreover be group-housed.
Telemetry can also enable reduction in animal numbers. Firstly, because telemetry systems are stable for months (and possibly even years), animals can be used as their own controls, reducing data variability and consequently the number of animals needed per treatment group. Secondly, telemetry provides an ability to continuously record a number of variables so that there is a significant increase in the amount of data that can be obtained from a given number of animals, compared to the use of conventional methods. Thirdly, in the absence of potential stressors, such as restraint or externalised catheters, the quality of data obtained is improved. Telemetry systems can also provide indicators of animal wellbeing to help implement earlier, more humane end-points. 2 Telemetry thus is widely regarded as benefiting science while minimising impact on animal welfare. However, the requirement for appropriate surgical training should not be underestimated, as this has a direct impact upon animal welfare. A sound basic and advance training in experimental surgery and good working knowledge of the devices are absolutely essential before progressing into implant insertion
Illustrative procedure
Study design
In this example, three pre-selected male beagle dogs (suitable temperament) will be used. Animals will act as their own control.
After overnight fasting, animals will be anesthetised. Telemetry devices allowing for continuous measurement of body temperature and cardiovascular parameters (ECG, blood pressure) will be surgically implanted. The implantation of these telemetric devices is a surgical procedure not invading body cavities, as the emitter is not implanted intraperitoneally, but in an inter-muscular pouch. The catheter is inserted in the femoral artery and further advanced into the aorta. ECG electrodes are tunnelled subcutaneously. The discomfort provoked is linked to the implantation and the surgical wounds created, and the animals’ need to be anaesthetised.
During the 3 week recovery period, animals will be trained to be socially isolated for 2 h (e.g. telemetric recording).
After recovery, the procedure calls for continued use: animals will be used weekly (one day with vehicle, and another with compound) to evaluate cardiovascular effects of the novel therapeutic agent at two different dose levels. The first treatment will consist of vehicle administration, followed by administration of the novel pharmaceutical agent. In-between recordings, animals will be socially housed. A minimal wash-out period will be allowed between test sessions (as determined by PK studies).
The study is intended to assess the potential cardiovascular effects of novel agents at proposed therapeutics doses.
At the end of the experiment, animals will be socially housed, while awaiting possible re-use.
Consideration of specific refinements and humane end-points

Initial prospective assessment
The procedure is classified as MODERATE as it requires anaesthesia and surgical intervention.
The administration of the compound is expected to have no clinical effect on the animal. However, administration of the compound is MILD, as it involves injection according to Good Veterinary Practice.
Could the severity classification be MILD?
No: the procedure involves anaesthesia and surgery; thus classification is MODERATE.
In the case of re-use of previously implanted animals that have been used in another project, the simple administration and evaluation through the use of telemetry of test compounds at therapeutic dose levels, and as no restraint is involved (freely moving) the prospective severity is to be classified as MILD
Could the severity classification be SEVERE?
Yes, if the compound tested at doses administered would have toxic effects (cardiovascular or other). This would definitely mean that the flow chart of testing is not accurate, as a model of telemetry is definitely not intended to evaluate toxic doses, but rather clinical/therapeutic doses of compounds. The study design would need to be reviewed.
Clinical observation/scoring system
Animals are carefully monitored at every step of the procedure. A specific anaesthesia sheet, as well as a clinical observation sheet, are developed, and included at the end of this example. During techniques, any clinical observation is recorded daily. During the wash-out periods, animals are observed and weighed at least weekly.
Results and assessment of actual severity
Three dogs underwent the procedure. All recovered from surgery without any complication, no cardiovascular side-effects were observed in any of the animals at any of the doses tested.
The three animals were considered to have experienced MODERATE severity.
At the end of the procedure the animals were again socially housed. The level of severity was MODERATE, due to the surgical implantation animals underwent, but the general state of health has been fully restored. No cumulative severity was observed due to administration of the novel therapeutic agent under evaluation. Therefore it is considered that animals may be re-used in future MILD, MODERATE or Non-recovery procedures pending an appropriate wash-out period and after positive veterinary advice.
Example clinical observation/scoring sheets
References
Evaluation of the efficacy of novel antiarrhythmic substances – rabbit model of atrial fibrillation using implanted mini-pump for administration of substances
General context
Paroxysmal atrial fibrillation (AF) is a condition in which an irregular heart rhythm occurs periodically. The heart returns to its normal sinus rhythm on its own in a few minutes, hours or days. People who have this type of AF may have episodes every day, or only a few times a year. When these episodes begin and end is usually unpredictable, which can be very unsettling. About 1 in 4 people with paroxysmal AF eventually develop the permanent form of the condition.
The causes of paroxysmal AF aren’t always known. Although AF can occur in patients without evident heart disease, organic heart diseases, such as congestive heart failure and mitral valve disease, are involved in about half the cases of paroxysmal AF and 80% of persistent or permanent AF. People with paroxysmal AF appear to be at just as high a risk of developing blood clots as those with chronic AF. However, the benefits of blood-thinning medications have not been shown to be as effective in people with paroxysmal AF as those with chronic AF. AF is a common arrhythmia and a potent risk factor for cardio-embolic stroke. There is evidence for many ion current changes in studies of the ionic properties from atrial tissue of patients with AF, and this is the reason that ion channel blockers require to be investigated.1,2
The development of numerous animal models of AF with clinically relevant disease paradigms has been a major advance over the past years. Models have been developed in rats, dogs, goats, sheep and more recently in genetically altered mice. 3 The severity and invasiveness of the model will depend on the underlying causes studied.
Screening based on electrophysiological, ionic, and molecular mechanisms in in vitro models allow identification of potential efficacy of compounds, which are then evaluated in in vivo models. The ideal animal model for drug testing should be simple, stable, inexpensive, clinically relevant, allow for reliable sustained AF production (for testing drug-induced termination) and eventually allow for easy conversion to sinus rhythm (for multiple dose studies). The example provided here concerns a surgical model which has proven good reproducibility in producing AF, together with minor clinical discomfort to the animal.
Illustrative procedure
Study design
In this example, 10 large adult male New Zealand White (NZW) rabbits (n = 5 per dose group (saline control and test substance)) will be used. Based on power analysis from previous data (preliminary studies) this was determined to be the minimal number of animals required per sample size.
In order to allow the test compound to be present as a preventive treatment in a steady state level, a subcutaneous mini-pump will be implanted which allows delivery of the compound for 21 days. This allows not only for continuous diffusion of the drug in the animal’s body (thus preventing peaks of drug distribution) but also minimises disturbance of the animal for drug administration (which can momentarily provoke supplementary changes in heart frequency and lead to spontaneous AF).
Under the same anaesthetic procedure, bipolar electrodes will be implanted on the left atrium and connected to an electrical stimulator. The electrical stimulator will be contained in a jacket. Animals will be habituated to the jacket before surgery.
A post -operative period of 7 days will allow the animals to fully recover from the surgery. Thereafter, implanted electrodes will be stimulated daily for 14 consecutive days. This allows a graded stimulus, gradually increasing heart rate and provoking paroxysmal AF. This will involve occasional handling and immobilisation of the animal in order to modify stimulation parameters.
14 days later, the animals will be terminally anaesthetised. Once hemodynamic parameters are stable, AF will be provoked by a fixed time period of electrical stimulation and hemodynamic parameters and biological markers will be measured.
Consideration of specific refinements and humane end-points

Initial prospective assessment
The study is intended to assess the efficacy of novel therapeutic agents in preventing/minimising occurrence of AF following electrical stimulation of the left atrium.
The s.c. implantation of a mini-pump is a very fast procedure; only takes a few minutes for an experienced person. The thoracic surgery is an invasive procedure. There is a need to anaesthetise the animal and the surgery may certainly provoke some pain and discomfort. The cardiac stimulation itself does not seem to provoke any pain, no visible discomfort: animals do not seem to react to increased heart frequency when this is done in a slow and progressive manner.
A prospective severity of MODERATE is deemed appropriate.
Could the severity classification be MILD?
No; the thoracic surgery will provoke pain and discomfort, which however is minimised by pain alleviation and best practice surgical techniques.
This procedure involves major surgery, even the implantation of the mini-pump alone would be a procedure which must be carried out under anaesthesia.
Could the severity classification be SEVERE?
If for whatever reasons no adequate analgesia or post-operative analgesia would be given this procedure could potentially provoke severe pain.
If cardiac stimulation would not be gradual, it might provoke spontaneous AF, which may lead to spontaneous death of the animal and thus should be classified as SEVERE. This is rare, and should with appropriate progressive stimulation only occur very rarely
Clinical observation/scoring system
Animals are very carefully monitored at every step of the procedure using a specific anaesthesia log and a clinical observation sheet. In the post-operative period, analgesia and local supportive therapy are provided as necessary and recorded on the individual clinical observation sheet. Sheets are filled out daily and include recordings of bodyweight, body temperature, clinical signs, standard post-operative treatments, any other treatments. Stimulations and reactions to it are also recorded. Examples of observation sheets are included at the end of this example
Results and assessment of actual severity
Ten animals underwent the procedure. All recovered from surgery without complications. One animal in the saline control group was found dead shortly after the first electrical stimulation. All other animals underwent the terminal anaesthesia during which efficacy of the therapeutic agent was evaluated versus vehicle. Severity assessment for the five saline-treated animals:
- 1 out of 5 rabbits was found dead at D11 (first electrical stimulation): SEVERE
- 4 out of 5 completed the terminal anaesthesia procedure: MODERATE
- 5 compound treated animals were used. All 5 completed the terminal anaesthesia procedure: MODERATE
Example of post-operative follow-up sheet
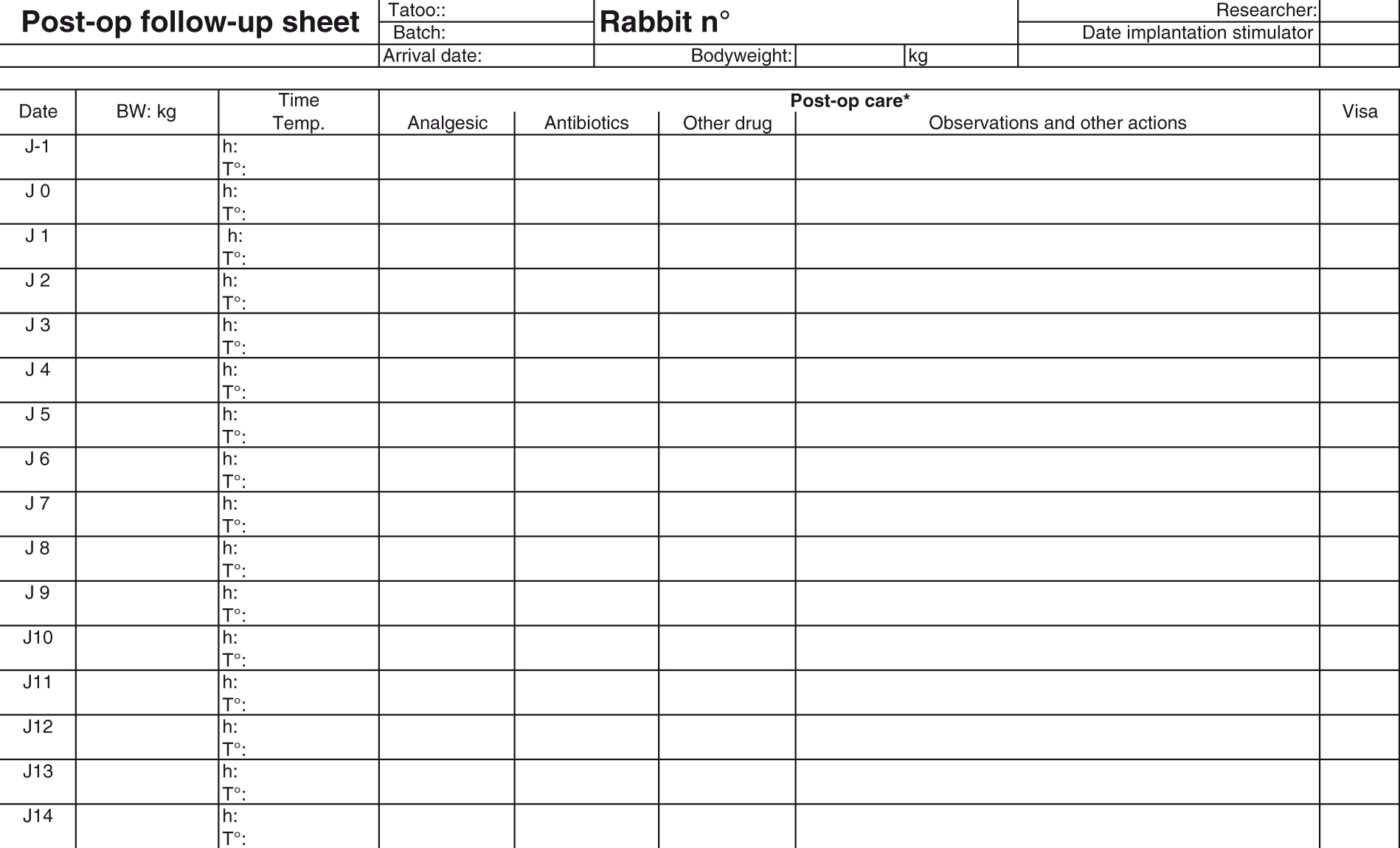
References
Determination of ecotoxicity of a test substance – bioconcentration flow-through fish test
General context
The bioconcentration test aims at determining the bioaccumulating potential of a chemical in tissues of fish, when triggered by (a.o.) its octanol-water partitioning coefficient. This is a legal requirement for all chemicals when exposure of the aquatic environment is possible. A study can be conducted with waterborne exposure; this will provide a bioconcentration factor (BCF, ratio of concentration in fish divided by concentration in water). The study may also be conducted with dietary exposure (e.g. when the substance is very poorly soluble in water); this will provide the biomagnification factor (BMF, ratio between concentration in fish divided by concentration in food).
The BCF or BMF is used in the assessment of the persistence, bioaccumulation and toxicity (PBT) and very persistent and very bioaccumulative (vPvB) criteria, and is used for the environmental risk assessment for the aquatic food chain (i.e. water – fish – top predators, such as birds and mammals). If a substance is bioaccumulative it will accumulate in living organisms once it enters the environment, which may lead to adverse effects higher in the food chain. A substance for which all criteria for PBT and/or vPvB are fulfilled will be identified as a PBT/vPvB substance. Substances identified as being PBT or vPvB are considered candidates for substitution, which means that they should be replaced by substances that are not PBT/vPvB if possible.
A PBT/vPvB assessment is required for all substances (i.e. industrial chemicals, agrochemicals, biocides and human and veterinary drugs). Though there are some QSARs available, these are not always suitable, depending on the molecular structure or physicochemical characteristics of the chemical. Furthermore, QSARs will only estimate the BCF of the unchanged chemical, while a study in fish allows for the determination of degradation products and elimination time upon transfer to uncontaminated water.
Fish species used in the test may involve: carp (Cyprinus carpio), zebra-fish (Danio rerio), fathead minnow (Pimephales promelas), bluegill (Lepomis macrochirus) or rainbow trout (Oncorhynchus mykiss).
Illustrative procedure
Study design
The study is performed based on the OECD guidelines for testing of chemicals, Guideline No. 305: Bioaccumulation in Fish: Aqueous and Dietary Exposure, October 2012. 1 The procedure is designed to meet the test methods prescribed by Commission Regulation (EC) No 440/2008 of 30 May 2008, Part C: Methods for the determination of ecotoxicity, Publication No. L142, C.13 ‘Bioconcentration: Flow-through Fish Test’. 2 The fish are exposed during an uptake phase of 28 days and a depuration phase of 56 days. The duration of both phases may be changed based on the course of uptake and/or depuration. During the uptake phase a group of fish is exposed to the test substance at one or more chosen concentrations. The numbers of fish per test concentration are selected such that a minimum of four fish are available at each sampling point. They are then transferred to a medium free of the test substance for the depuration phase. The fish are kept in tanks with a continuous supply of water, to which the test substance is added during the uptake phase. The water volume should be replaced at least five times per day. Non-toxic concentrations of test substances are used. Radiolabelling of the test substance may be used for analytical purposes. Water samples are taken prior to and during uptake and depuration phase. Fish are selected for tissue sampling following euthanasia at sequential time points during the uptake phase (e.g. 1, 3, 7, 14, 21 and 28 days) and the depuration phase (30, 35, 42, 56 and 84 days); and for lipid extraction at 0 or 28 days of exposure. Weighing is performed before test and post-mortem; sampling of fish during test and post-mortem. Four or more fish are needed per sampling point and per test concentration. A total number of 130 rainbow trout were used and exposed to two different concentrations of a test substance: 32 fishes will be used for the control group (1 replicate) and 49 fishes for each test concentration (2 replicates).
Consideration of specific refinements and humane end-points

Initial prospective assessment
Animals are only expected to experience MILD discomfort based on exposure to non-toxic concentrations of test substance, and the use of good practice for handling and euthanasia.
A prospective severity classification of MILD is therefore appropriate.
Could the severity be MODERATE?
Normally not, as during the bioaccumulation phase only slight or no adverse effects, and thus only MILD discomfort are expected based on use of non-toxic dose concentrations, good practice handling and euthanasia methods. If contrary to expectations clinical signs indicating moderate severity are observed, an informed decision is made on the follow-up of the experiment, e.g. euthanasia of the animal based on humane end-points, intensified monitoring or need for change of prospective severity assessment of similar experiments and notification to CA.
Clinical observation/scoring system
Animals are monitored for mortality/viability and any adverse effects daily. Any clinical score sheet and observation procedure should be agreed upon by the researcher, veterinarian and animal technologists to set up criteria for monitoring and timely euthanasia (humane end-points).3–5 Temporary use of light to improve observations is possible but should be limited. Fish are observed for adverse effects and mortality after 2–4 h, and for mortality or possibly more adverse effects for possible implementation of humane end-points at the end of the day. Then following mornings, the fish are checked for any mortality, and for mortality and any (more) adverse effects on the observation moments, and at the end of the day. An example of the clinical observation and scoring system used to help monitor the clinical condition of animals throughout the procedure is included at the end of the example. Unless typical clinical signs or abnormalities (e.g. typical swimming behaviour or damaged fin/tail) are present in one individual or very small number of fish, any follow-up or re-identification of an individual fish is not or hardly possible. Whenever it is possible to identify an individual affected fish with subsequent observations, this may be helpful in monitoring the clinical condition, appropriate for possible implementation of humane end-points. If not, counting of the number of fish with the typical clinical signs should be used to assess the condition of the group of fish per tank.
Results and assessment of actual severity
During the 84 days, 124 fish did not show any clinical signs and were considered to have experienced MILD severity. In test concentration 2, discolouration was observed in one fish on three consecutive days, followed by complete recovery. This was likely to be the same fish on each day. Severity was considered MILD. In the control group, test concentration 1 and 2, one fish was found dead without prior clinical signs. Severity was considered MILD. In both the control group and test concentration 1, one fish was circle swimming. After consultation of the designated veterinarian, these fish were kept in study and closely monitored. Clinical signs disappeared in the fish after two days. Severity in both fish was considered MODERATE. Finally, in test concentration 1, one fish was observed without a tail and humanely euthanised. Severity: SEVERE.
Assessment: MILD for 127 fish, MODERATE for 2 and SEVERE for 1 fish.
Example clinical observation/scoring system
A clinical score sheet can be used for daily observations on mortality and adverse effects. A clinical score sheet may include several signs regarding swimming behaviour, pigmentation, appearance, reactive behaviour or convulsions. Their presence or absence is documented daily or more frequently is necessary. Any abnormal and unexpected behaviour should be reported to the researcher and/or designated veterinarian or suitably qualified fish expert. If severe adverse effects are observed, it should be decided to monitor more frequently and/or to implement humane end-points. Considering the absence of identification and group density, follow-up on any observations on individual animals may be impaired or non-realistic.
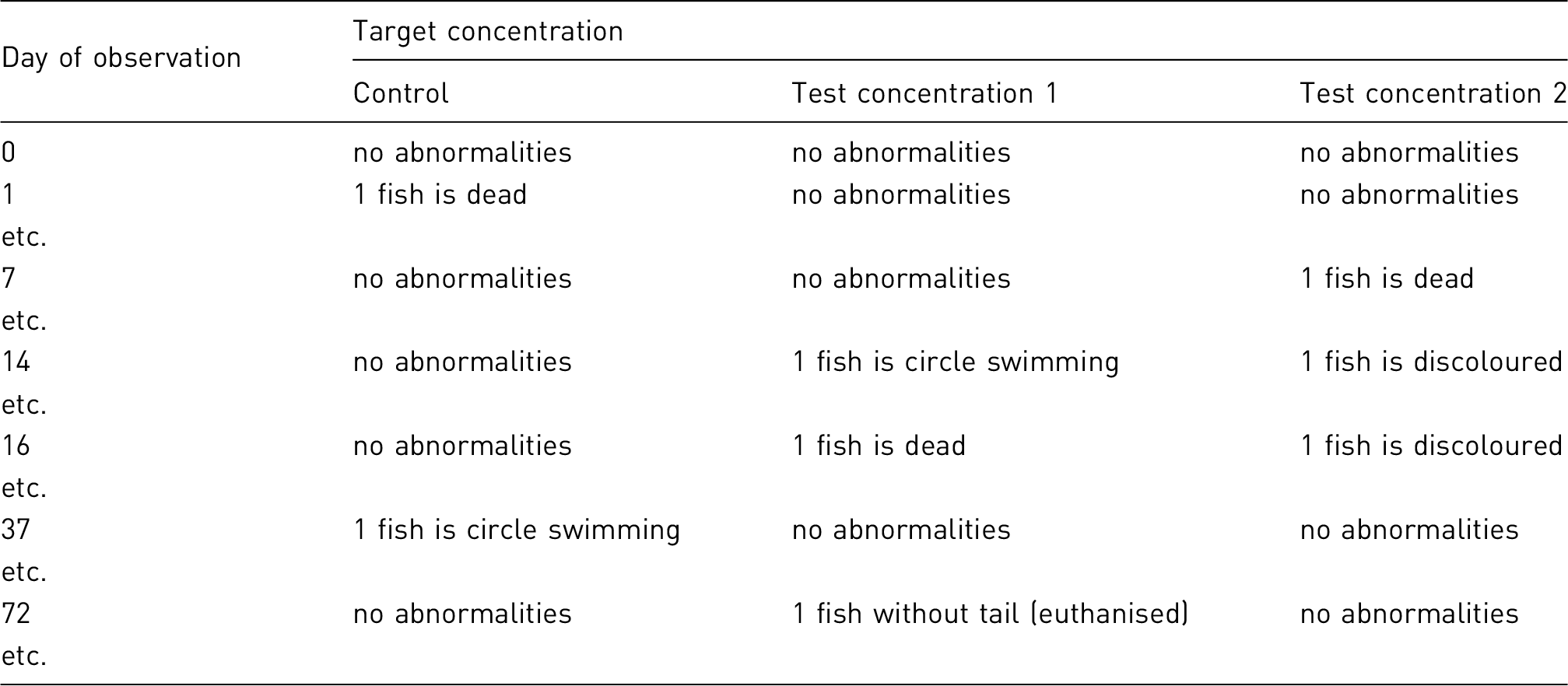
Severity assessment is performed by a combination of these observations (together with a procedure for evaluation). In case of combinations of observations as described in the table, the highest score present is used for assessment. If an observation is present for more than one day in a row, the scores may be adjusted (+1, +2 or +3) in consultation with the researcher, animal technologists and/or designated veterinarian or fish expert. Close monitoring is helpful in the assessment of actual severity on MILD, MODERATE or SEVERE, especially in cases of sudden death with or without previous clinical signs or in helping taking informative decisions on timely implementation of humane end-points.
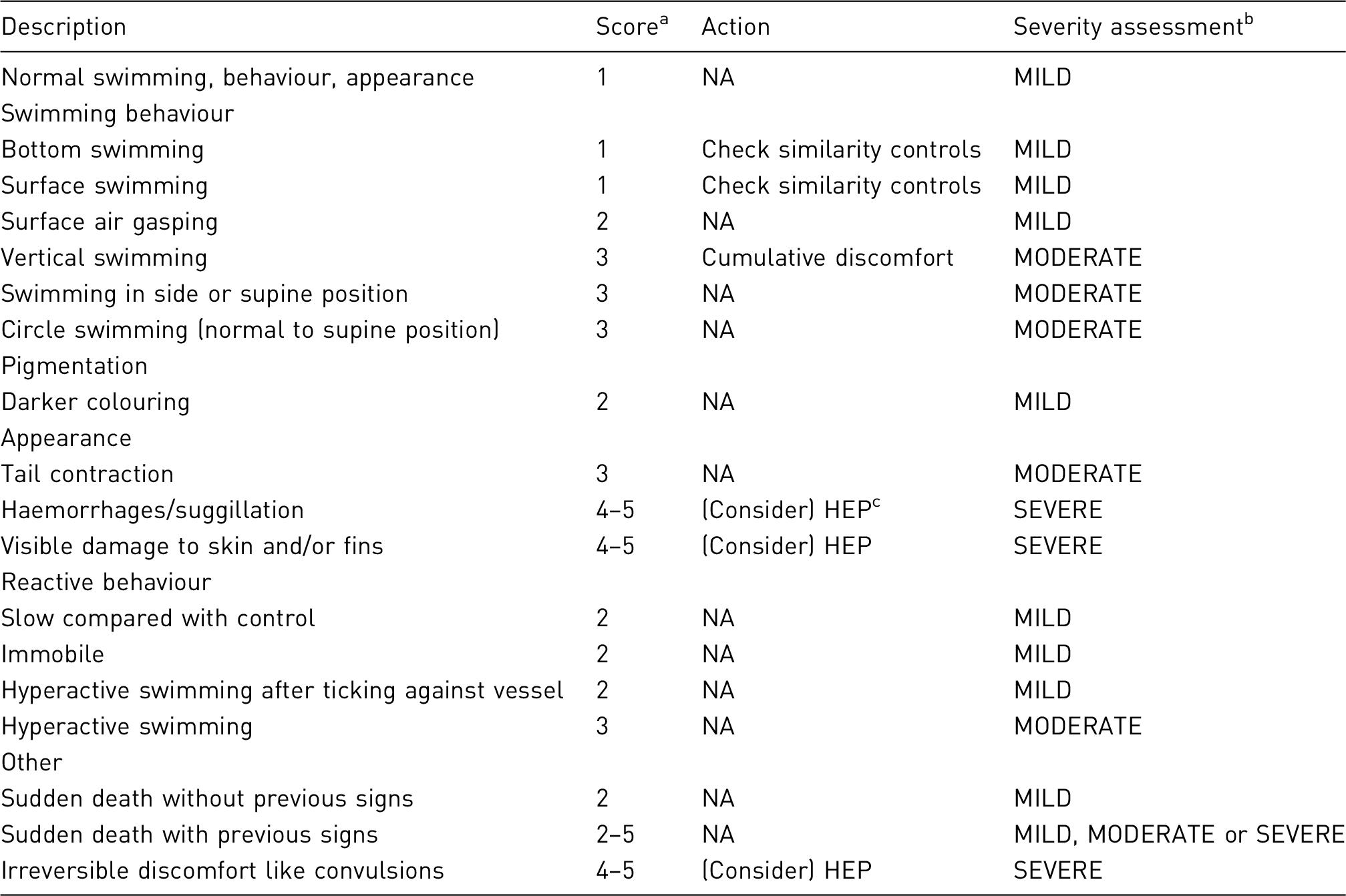
If an observation is also present on the second or third day in a row, the score is added +1, 4 days in a row +2 and 5 days in a row +3. This may facilitate severity assessment and implementation of HEP.
If a combination of observations is present, the severity assessment will be adjusted to the highest severity score of the observations present.
HEP = humane end-points.
References
Assessment of acute oral toxicity with a test substance in rats
General context
The Organisation for Economic Co-operation and Development (OECD) test guidelines are a collection of methods used to assess the hazards of chemicals and of chemical preparations such as pesticides. These methods cover tests for physical and chemical properties, effects on human health and wildlife, and accumulation and degradation in the environment. The OECD test guidelines are recognised worldwide as the standard reference tool for chemical testing and are periodically reviewed in the light of scientific progress or changing assessment practices. Acute oral toxicity data are used to satisfy hazard classification and labelling requirements, for risk assessment for human health and the environment, and when estimating the toxicity of mixtures. The objective of an
The conventional acute oral toxicity test (formerly OECD Test Guideline 401, the classic ‘LD50’ test), 1 requiring more than 20 animals is the most heavily criticised test in terms of animal welfare and this concern was the driving force behind its deletion and the development of three alternative tests for acute oral toxicity (OECD test guidelines 420, 423 and 425),2–4 respectively, the fixed dose method (420), the acute toxic class method (423), and the up-and-down procedure (425). All guideline tests involve giving the test substance in graduated doses to groups of animals, which are observed with respect to effects and deaths (or humane end-points). The result is given as a calculated LD50 (the dose that kills 50% of the animals) or range estimates of LD50. The fixed dose method does not require the death of animals as an end-point and uses on average 5–7 animals with 1 death. The acute toxic class and up-and-down procedure use on average 6–9 animals, with 0–3 deaths. 5
Illustrative procedure
Study design
The acute toxic class method is a stepwise procedure with the use of three animals of the most susceptible sex (if unknown, female) per step. All available information on the test substance (e.g. identity and chemical structure, physico-chemical properties, other in vivo or in vitro toxicity tests, toxicological data on the structurally related substances, anticipated use(s) of the substance) should be considered prior to conducting the study.
The test substance should be formulated using a suitable vehicle and concentrations should be adjusted to allow for constant dosage volumes. Animals should be fasted prior to dosing to avoid interference with the test substance by food present in the stomach; the guideline requires ‘overnight’ fasting. Dosing volumes are chosen according to good practice and vehicles according to internal/external recipients databases.
The dose level to be used as the starting dose is selected from one of four fixed levels (5, 50, 300 and 2000 mg/kg). There is an option to use an additional dose level of 5000 mg/kg, but only when justified by a specific regulatory need. The starting dose level should be that which is
The time interval between treatment groups will be based on onset, duration and severity of the toxic signs. Treatment of next group will be delayed until no further mortality is expected in previously dosed group(s). On average 2–4 steps may be necessary to enable a judgement with respect to classifying the test substance to one of a series of toxicity classes defined by fixed LD50 cut-off values.
In total, six rats will be used in this study. According to the stepwise procedure, in step one, three rats will be dosed by oral gavage at 300 mg/kg as a starting dose. Animals will receive a single oral dosing and then be observed for 14 days. Observations include clinical signs, body weight measurements and macroscopic abnormalities during necropsy at the end of the observation period, or earlier if found dead or humanely killed.
Consideration of specific refinements and humane end-points

Initial prospective assessment
SEVERE: Substance toxicity may result in up to
In general, a severity classification of
Yes, if test substance information suggests mortality to be unlikely at the highest starting dose level (2000 mg/kg BW) and mortality or any relevant (delayed) clinical signs or mortality remain absent in 6 rats (3 animals per step), the severity may be considered
Clinical observation
According to the guideline (OECD TG 423), observations should be made in individual rats after dosing at least once during the first 30 min, then periodically during the first 24 h, with special attention given the first 4 h, and daily thereafter for 14 days. 3 Duration of observation should be determined by toxic reactions, time of onset and length of recovery period and may be extended when considered necessary. Body weights are measured before dosing and at least weekly thereafter. Body weight changes should be recorded and used for welfare assessment and/or early humane end-points. Any relevant macroscopic abnormalities during necropsy may be useful to confirm the correctness of chosen humane end-points. Additional observations may be necessary if signs of toxicity continue. Observations should include changes in skin and fur, eyes and mucous membranes, and also respiratory, circulatory, autonomic and central nervous systems, and somatomotor activity and behaviour pattern. Attention should be directed to observations of tremors, convulsions, salivation, diarrhoea, lethargy, sleep and coma. The principles and criteria summarised in the OECD Humane Endpoints Guidance Document should be taken into consideration. 6 Animals found in a moribund condition and animals showing severe pain or enduring signs of severe distress should be humanely killed.
A clinical score sheet or program (FELASA Working Group Report, 2015) is useful for monitoring animal welfare, identifying humane end-points as well as reporting according to the OCDE guidelines.6,7 An example of a clinical scoring system which is used to help monitor the clinical condition of the animals throughout the procedure is included at the end of this example.
Results and assessment of actual severity
All treated rats in step 1 at starting dose of 300 mg/kg showed clinical signs. Clinical signs comprised lethargy, hunched posture, uncoordinated movements, piloerection, ptosis, rales and/or salivation on Days 1 and 2 where one rat showed hunched posture also on days 6 and 7. No rats were found dead or needed to be killed humanely, thus a second step was necessary according to the guidelines. In step 2, with 3 rats dosed at 2000 mg/kg, one female rat was found dead on Day 2, showing slight body weight loss and clinical signs like lethargy, hunched posture, piloerection, ptosis and salivation between Days 1 and 2. No macroscopic abnormalities were found during necropsy of this animal and other rats at the end of the study. No mortality was observed in other rats. Body weight gain of surviving rats was comparable to untreated animals of same strain and age. For this purpose, historical data on untreated or on same vehicle treated animals of same strain and age may be used.
Scoring system and examples of scoring sheets
Severity assessment is performed by a combination of clinical observations (changes in bodyweight, skin, fur, eyes and mucous membranes, occurrence of secretions and secretions and respiratory, circulatory, autonomic and central nervous systems, somatomotor activity and behaviour pattern.) based on a clinical scoring system used in toxicology, and necropsy findings. Time of onset, grade and duration of observed signs are recorded. Signs are graded for severity and the maximum grade is predefined at 3 or 4. Grades are coded as slight (grade 1), moderate (grade 2), severe (grade 3) and very severe (grade 4). For certain signs, only its presence (grade 1) or absence (grade 0) is scored.
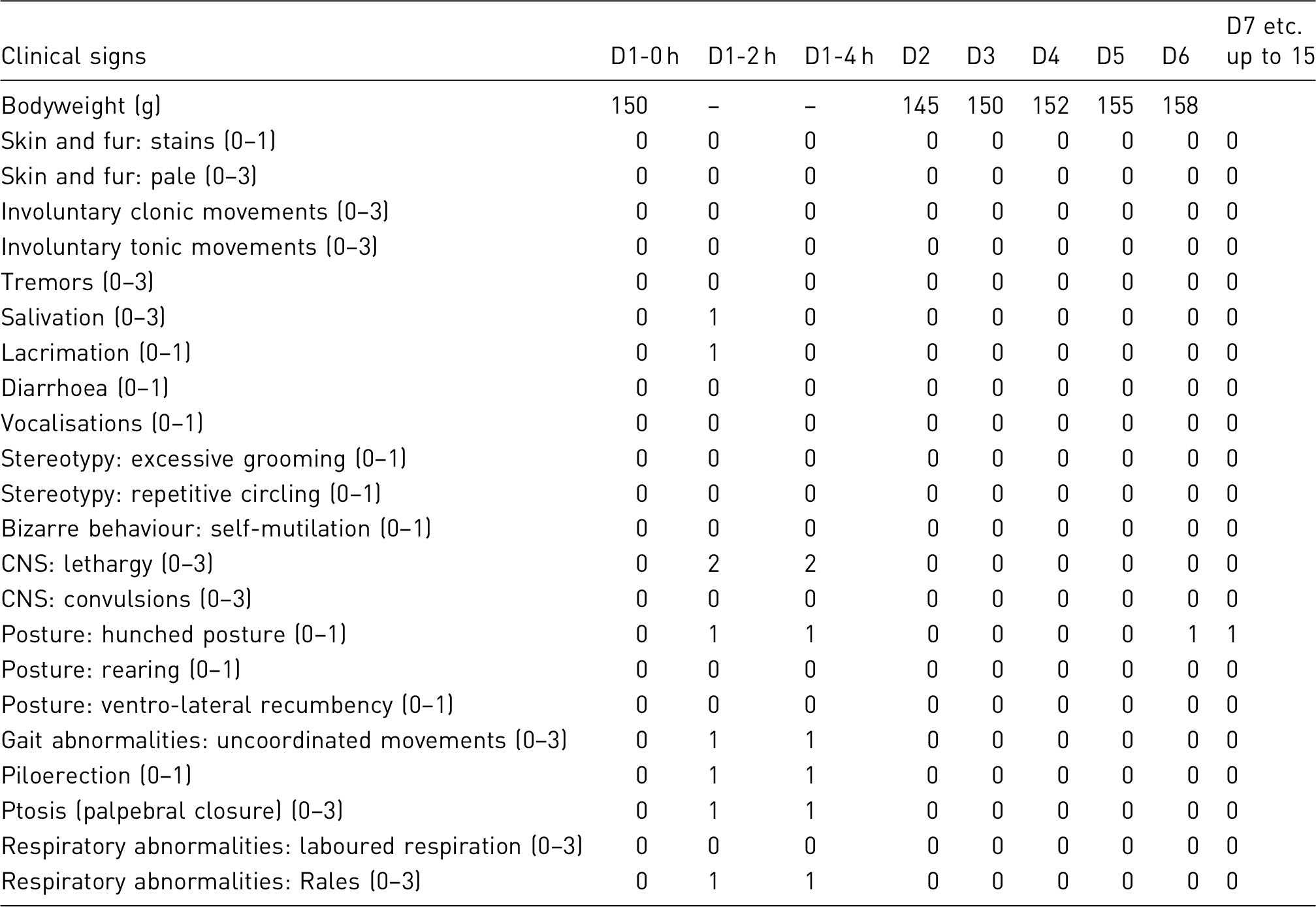
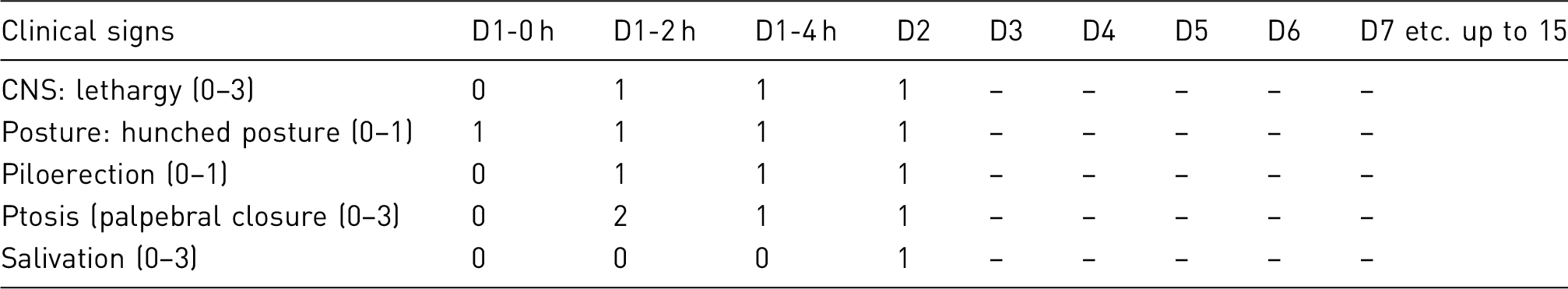
References
Pharmacokinetic study after single administration of a test substance in the dog
General context
The aim of the study is to study the pharmacokinetics of a compound after single administration. The difference in systemic exposure following oral (capsule/tablet/gavage/diet), intravenous, dermal or subcutaneous (bioequivalence) administration is assessed. For substances intended to be ingested via the oral route in humans, the oral route is compared to the intravenous route, which is the reference route giving 100% bioavailability. The applicable testing guidelines for the compound relate to any human exposure, prospective use or legislation.1–10 The compound may concern pharmaceuticals for human or veterinary use, feeding ingredients or (agro-)chemicals.
Illustrative procedure
Study design
A common study design is to use the same animals for two periods. In this example, three dogs were used. In the first period, plasma levels of the compound following an intravenous dose are measured, then after an appropriate estimated wash-out, in the second period, plasma levels of the compound following oral dosing of the same animals are measured. Third or more periods with appropriate wash-outs may be used to compare different oral test substance formulations (e.g. different solutions, tablets, capsules). For bioequivalence studies where a test compound is compared to a known compound, a randomised block design may be used to randomise the order of exposure to different routes and thus prevent any possible time effects from previous dosing. For bioavailability studies, where an unknown test compound is tested for sufficient bioavailability in order to be used for further studies, this may not be typically required. Urine and/or faeces may be collected from animals kept in metabolism cages for the collection. After the study, it may still be scientifically plausible to re-use the animal, and it may be allocated to the ‘stock animals’. Fitness for possible re-use will be based on age, lifetime and procedure-specific welfare assessment, restoration of general health and well-being in accordance with veterinary advice and classification of further procedures. A total of 4 periods, first period of intravenous dosing followed by three periods of different capsule dosing, each with a 1 week wash-out period in between the dosing periods, were used in the study. Observations comprised clinical signs and body weight measurement before each dosing period.
Consideration of specific refinements and humane end-points

Initial prospective analysis of severity
Animal is only expected to experience MILD discomfort based on limited food deprivation/water restriction, limited individual housing, single dosing, and repeated blood sampling. Test substance effects are expected to be absent or very limited due to levels considered pharmacological.
Single dosing of test substance and frequent blood sampling in the dog is considered to be MILD based on species character/behaviour (social interaction with humans), restraining method, and best practice dosing & sampling technique/volumes used. MODERATE severity seems unlikely prospectively as not toxic, but pharmacologic, doses are to be used, but may be caused by unexpected test substance (or vehicle) effects or possible complications during dosing and sampling.
Clinical observations
Veterinary examinations are performed before start of study (at arrival from the supplier or at periodic intervals in the animal stock) to ensure the good state of health and fitness for (re-)use of the animal for the procedure. Body weights are measured before each dosing period. When considered necessary, veterinary examinations may be performed during the study. During the study period, animals are monitored on mortality/viability at least twice daily and for clinical signs during dosing and the follow-up period at least once daily. 12

Severity assessment is performed by a combination of general clinical observations (body weight, appearance, behaviour, cage environment) based on a clinical scoring system used in toxicology. Time of onset, grade and duration of observed signs are recorded. Signs are graded for severity and the maximum grade is predefined at 3 or 4. Grades are coded as slight (grade 1), moderate (grade 2), severe (grade 3) and very severe (grade 4). For certain signs, only its presence (grade 1) or absence (grade 0) are scored.
Results and assessment of actual severity
In the first period, no mortality occurred. In the first period of intravenous dosing, one animal (no. 3 in the example) showed clinical signs like slight/moderate tremor, slight lethargy, hunched posture, vomiting of food and mucus, faeces containing mucus, and/or faeces with red particles for a period of 2 h, followed by a quick and complete recovery.
The mild but nevertheless clear clinical signs of relatively short duration after IV dosing could indicate similar signs for a longer duration after oral dosing. Therefore, the prospective assessment for periods of oral dosing was upgraded to MODERATE. Before continuation of the study, the CA was notified on the upgrade of the prospective assessment.
In the second (oral dosing) period, the animals showed no clinical signs.
Examples of clinical score sheets
Example of clinical scores in animal 1:
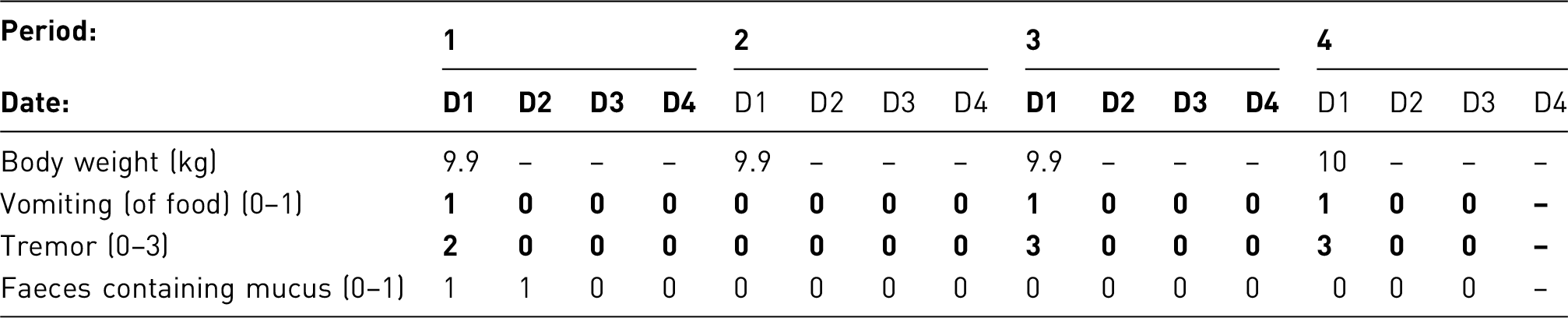
Example of clinical scores in animal 3:
Example of clinical scores in animal 3:
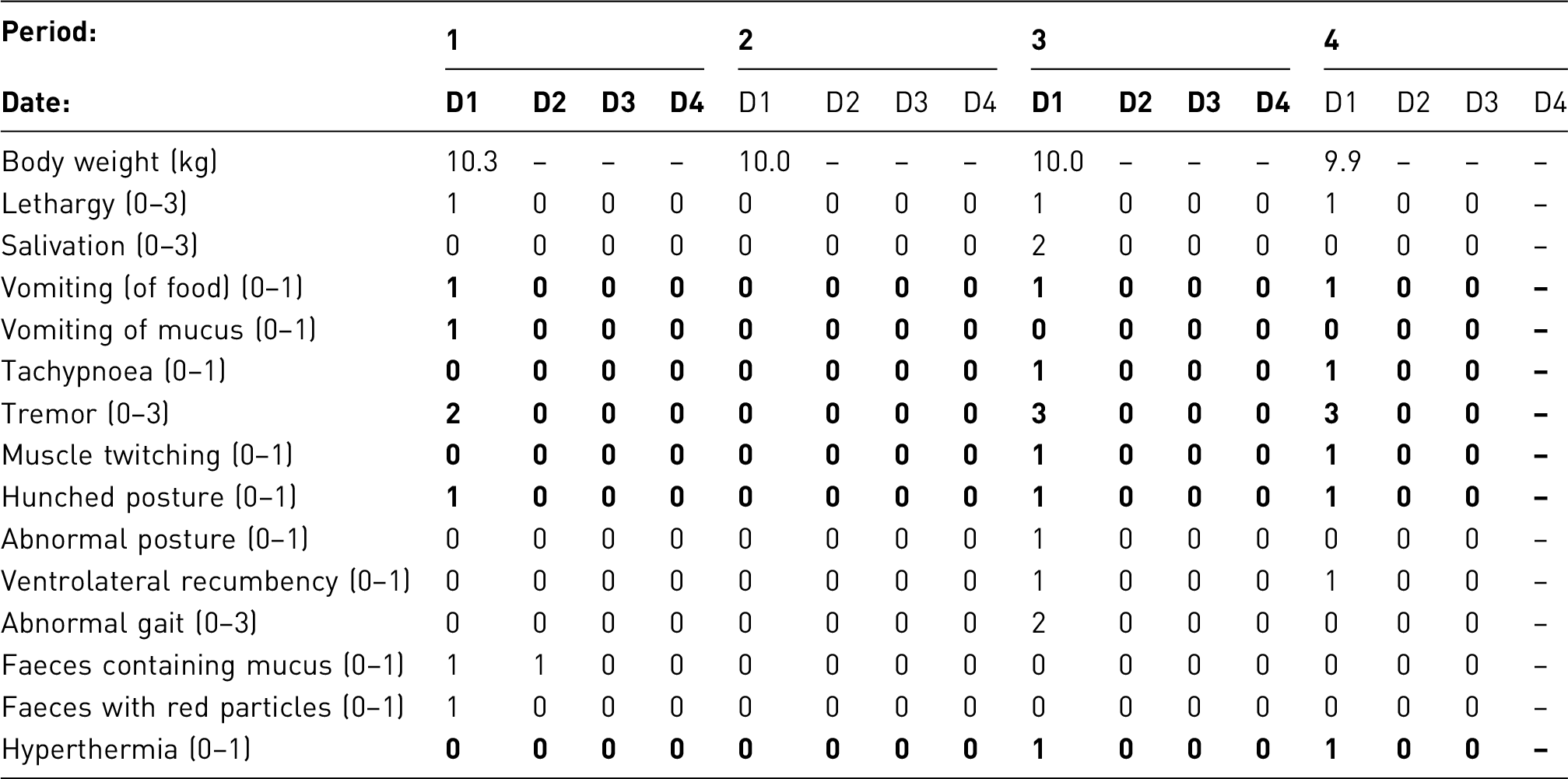
Assessment of actual severity in the three examples
Cumulatively over the four dosing periods, during actual severity assessment, two animals were considered as
Actions
As clinical signs are usually noted just after (IV) and/or during the first day after dosing (PO), closer monitoring may be essential in case of unexpected moderate to severe adverse effects, supported if necessary by veterinary consultation, notification to CA before continuation and any intervention like humane killing in case of humane end-points. 13 If moderate to severe adverse effects are noted in the first animal, a staggered start (some time interval between first and second or each animal, with consideration of expected duration of possible clinical signs) and reconsideration of the dosage is considered good practice. Also any possible differences between sexes need to be considered before the study or, if unknown, in the study design (e.g. starting with one animal per sex, then following animals of each sex).
After the last dosing and observation period, in case of mild up to moderate effects, the dogs are released from the study and monitored in stock until no further abnormalities are observed. In case of any planned re-use, it should be demonstrated that the general health and well-being of the animal has been fully restored until use for any next study will be considered. This assessment should be in accordance with veterinary advice and preferably supported by periodic clinical and laboratory examinations. If necessary, based on these findings and bioanalysis data (elimination half-time determined) in the study, an appropriate wash-out period between studies should be considered.
References
Supplemental Material
Suppplemental Material1 - Supplemental material for Classification and reporting of severity experienced by animals used in scientific procedures: FELASA/ECLAM/ESLAV Working Group report
Supplemental material, Suppplemental Material1 for Classification and reporting of severity experienced by animals used in scientific procedures: FELASA/ECLAM/ESLAV Working Group report by David Smith, David Anderson, Anne-Dominique Degryse, Carla Bol, Ana Criado, Alessia Ferrara, Nuno Henrique Franco, Istvan Gyertyan, Jose M Orellana, Grete Ostergaard, Orsolya Varga and Hanna-Marja Voipio in Laboratory Animals
Supplemental Material
Suppplemental Material2 - Supplemental material for Classification and reporting of severity experienced by animals used in scientific procedures: FELASA/ECLAM/ESLAV Working Group report
Supplemental material, Suppplemental Material2 for Classification and reporting of severity experienced by animals used in scientific procedures: FELASA/ECLAM/ESLAV Working Group report by David Smith, David Anderson, Anne-Dominique Degryse, Carla Bol, Ana Criado, Alessia Ferrara, Nuno Henrique Franco, Istvan Gyertyan, Jose M Orellana, Grete Ostergaard, Orsolya Varga and Hanna-Marja Voipio in Laboratory Animals
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.