
Abstract
Golden Syrian hamster embryos are difficult to cryopreserve due to their high sensitivity to cryoprotectants and in vitro handling. The objective of this study is to develop a robust open pulled straw (OPS) vitrification technique for cryopreserving hamster embryos at various developmental stages. We first systematically tested the concentrations of cryoprotectants and the exposure times of two-cell embryos to various vitrification solutions. We identified pretreatment of two-cell embryos with 10% (v/v) ethylene glycol (EG) + 10% (v/v) dimethylsulfoxide (DMSO) for 30 s followed by exposure in the vitrification solution, EDFS30 (containing 15% EG + 15% DMSO), for 30 s before plunging into liquid nitrogen (two-step exposure method) as the optimal OPS vitrification protocol. We then investigated the resourcefulness of this protocol for vitrifying hamster embryos at different developmental stages. The results showed that high blastocyst rates from embryos vitrified at two-cell, four-cell, eight-cell, or morula stage (62%, 78%, 80%, or 72%, respectively), but not those verified at pronuclear (0%) or blastocyst stage (24%; P < 0.05), were achieved by this protocol. When embryos vitrified at the two-cell stage were recovered and then directly transferred to recipient females, 29% of them developed to term, a development rate not significantly different (P > 0.05) from the 40% birth rate of the unvitrified controls. In conclusion, we have developed an effective two-step OPS vitrification protocol for hamster embryos.
Keywords
The golden Syrian hamster (Mesocricetus auratus) has been used as an animal model in many research areas, including virology,1–3 endocrinology, 4 metabolism,5,6 diabetes, 7 cancer,8,9 and cardiovascular diseases. 10 With the recent completion of the draft assembly of its genome by the Genome 10 K Projects at the National Human Genome Research Institute (NHGRI: www.genome.gov), the application of hamsters as animal models for biomedical research will certainly increase. Furthermore, the recent technology advancement achieved by us in employing the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9 (CRISPR/Cas9) system to efficiently conduct gene targeting in hamsters 11 has overcome another major barrier set up by the lack of gene targeting tools in this species. Other researchers are also making progress in developing tools to genetically modify the hamster genome. 12 Such technology advancement will inevitably promote the production and application of genetically-engineered hamsters in the near future. Since the maintenance of transgenic and gene-modified lines is costly and takes up space in animal facilities, cryopreservation of hamster embryos will be widely used and may become a routine in the laboratory. However, a simple and reliable cryopreservation technique for cryopreserving hamster embryos is currently lacking.
In 1985, Ridha and Dukelow 13 reported the first cryopreservation of hamster preimplantation embryos with slow freezing, but failed to produce live offspring. It is difficult to cryopreserve hamster embryos for several reasons. First, the widely used vitrification cryoprotectants, dimethylsulfoxide (DMSO) and ethylene glycol (EG), have detrimental effects on the in vitro development of earlier stage hamster embryos. 14 Second, hamster embryos are extremely sensitive to environmental factors, including light, 15 and chemicals,16,17 as well as the duration and temperature of culture medium equilibration, 18 which are normally tolerated well by embryos from other species. In agreement with these, Mochida et al. showed that only eight-cell stage hamster embryos could be cryopreserved and that the cryopreserved embryos had very poor chances of development into blastocysts after being frozen and thawed. 14 So far, only one group has reported successful vitrification of hamster embryos with a cryoloop method and, with a relatively lower level of success, with an open pulled straw (OPS) method. 19 In this study, only one-cell and two-cell stage hamster embryos were used. 19 While the cryoloop technique seems to be adequate for cryopreserving one-cell and two-cell stage hamster embryos, it requires direct contact between embryos and liquid nitrogen (LN2), 20 which raises concerns about the risks of contaminating embryos with infectious pathogens from LN2. 21
OPS vitrification was developed to cryopreserve bovine oocytes and in vitro produced embryos at various developmental stages by Vajta et al. in 1998. 22 Subsequently, studies have reported on the successful application of OPS techniques in cryopreserving mouse,23,24 rat, 25 rabbit, 26 sheep, 27 goat, 28 and porcine 29 embryos. Both OPS and cryoloop vitrification, using a minimum volume of cryoprotectants, belong to the third generation of cryopreservation technology, 30 relative to the slow freezing and conventional vitrification methods. The aims in using the OPS method are to prevent ice formation and reduce chilling injury, toxicity of cryoprotectants and osmotic damage to embryos through improving cooling and warming rates. 22 Many reports have shown that the OPS method is superior to slow freezing with regard to survival, in vitro and in vivo development of cryopreserved embryos.26,31,32 Moreover, by applying a modified procedure for embryo loading, the OPS method can be performed under sterile conditions and can comply with sanitary regulations.33,34
The present study was designed to establish and optimize an OPS method for hamster embryo cryopreservation and to investigate the effectiveness of such a method in cryopreserving embryos at different developmental stages.
Materials and methods
Animals
Golden Syrian hamsters used for embryo collection were bred in-house by using founder animals purchased from Charles River, Kingson, NY, USA (LVG Golden Syrian Hamster, Strain Code: 049). Black Syrian hamsters used as recipients for embryo transfer were acquired from a breeding colony established in our laboratory. All hamsters were raised and maintained in an air-conditioned room with a 14:10 light–dark cycle (lights on from 06:00 h). The hamsters used were healthy and free of rodent pathogens, as demonstrated by routine monitoring of sentinel animals housed in the same room. In total, 90 golden Syrian females were used as embryo donors and 25 black females as recipients for embryo transfer. The experiments were conducted in accordance with the guidelines of the Laboratory Animal Research Center at the Utah State University and approved by the Institutional Animal Care and Use Committee (IACUC Protocol: 2091).
Reagents and media
All reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. Hamster embryos were cultured in HECM-916 supplemented with 0.5 mg/mL human serum albumin (A1653). The Hepes-buffered HECM-9 (HHECM-9) was used for in vitro manipulation of embryos, which contained 20 mmol/L HEPES and 5 mmol/L NaHCO3. The pretreatment solution for embryo vitrification consisted of 10% (v/v) EG+10% (v/v) DMSO in HHECM-9. Four vitrification solutions consisted of 12.5% EG+12.5% DMSO, 15% EG + 15% DMSO, 17.5% EG + 17.5% DMSO, and 20% EG + 20% DMSO in HHECM-9 supplemented with 300 mg/mL Ficoll and 171.2 mg/mL sucrose, defined hereafter as EDFS25, EDFS30, EDFS35, and EDFS40, respectively.
Embryo collection
Hamster embryos were collected from 2–3-month-old, cycling golden hamsters. Hamsters were induced to superovulate by intraperitoneal injection of pregnant mare’s serum gonadotrophin (PMSG, G4877) with weight-dependent dosages (10 IU if <90 g, 15 IU if 91–115 g, 20 IU if 116–140 g, and 25 IU if >141 g) 16 at 09:00 h on the day of post-estrus discharge (day 1 of cycle). 35 Females were mated to fertile males at 19:00 h on day 4. One-cell embryos at the pronuclear (PN) stage were collected from oviducts at 15:00 h on day 1, two-cell at 08:00 h and four-cell at 22:00 h from oviducts on day 2, eight-cell at 16:00 h and morula at 20:00 h from uteri at day 3, and blastocysts at 08:00 h from uteri at day 4, by flushing with HHECM-9 pre-equilibrated in a CO2 incubator (37.5℃, 10% CO2, 5% O2, and 85% N2). Shortly after collection, embryos at different stages were either cultured to blastocysts or subjected to vitrification. The in vitro manipulation of embryos was performed in a dark room with a small incandescent lamp, and red filters were used on the microscope light source, as described by Yamauchi et al. 36
OPS vitrification
The OPS was made using the method described by Vajta et al. 22 with some modifications. Briefly, a 0.25 mL straw (IVM, L’Aigle, France) was heat-softened over a small alcohol burner, pulled manually and cut at the tapered end with a blade. The inner diameter of the tip was 0.20–0.25 mm and the wall thickness was approximately 0.05 mm. Embryo freezing and thawing were performed on a 37.5℃ hot plate, and the ambient temperature was maintained at 25 ± 1℃. Embryos were pretreated in 10% EG + 10% DMSO for 30 s, and then transferred to the EDFS solution and held for 30 s, 60 s, or 120 s, respectively. After that, embryos were picked up by the narrow end of the OPS. The OPS loaded with embryos was immediately plunged into LN2. Approximate 10 embryos were loaded into each OPS and stored in LN2 for at least 48 h. When thawing, the OPS was taken out of the LN2, and the narrow tip was immersed immediately in 1 mL of warmed (37.5℃) 0.5 M sucrose. Embryos were expelled from the OPS and transferred into another drop of the same solution for 5 min to dilute cryoprotectants. After thawing, embryos were either cultured to blastocysts or transferred to recipients after 0.5 h recovery in HECM-9.
Embryo culture
Embryos were placed into 20 µL drops of HECM-9 covered by mineral oil in groups of 8–15 and cultured at 37.5℃ in a humidified atmosphere of 10% CO2, 5% O2, and 85% N2. Culture dishes were pre-equilibrated for at least 5 h in an incubator before use.
Differential immunocytochemical staining
Embryos were fixed and extracted with 4% paraformaldehyde in phosphate-buffered saline (PBS) containing 0.5% Triton X-100, 1 mmol/L dithiothreitol and 0.3% bovine serum albumin (BSA) overnight at 4℃. After washing twice with PBS containing 0.01% Triton X-100, embryos were blocked in PBS containing 150 mmol/L glycine, 0.02% sodium azide, 0.1% Triton X-100 and 2% BSA for one hour. The embryos were then incubated in goat anti-sox 2 antibody (Santa Cruz Biotechnology, CA, USA) (1:100) in PBS for one hour. They were washed with 0.05% Tween 20 and incubated in Alexa Fluor 594 donkey anti-goat IgG (Invitrogen, Carlsbad, CA, USA) (1:500) for 30 min. DNA was stained with 20 µg/mL of Hoechst 33342 for 20 min. Finally, embryos were mounted on slides in 50% glycerol in PBS and then examined under a Zeiss epifluorescent microscope (Carl Zeiss Optical, Inc, Chester, VA, USA). Images were captured by digital camera with Axio Vision LE Program (Carl Zeiss, Thornwood, NY, USA).
Embryo transfer
Viability of vitrified two-cell embryos was assessed by transfer to recipient females. Black Syrian females, naturally mated with black males, one day before, were used as recipients, and the color of hair was used to distinguish between pups from transferred embryos and from natural mating. The surgery of embryo transfer was based on the procedure described by Farrell and Bavister 37 with some modification. The recipients were induced to anesthesia by intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine. Approximately 2 cm incisions were made with fine dissection scissors on the left and right paralumbar sites. After pulling out the ovary, oviduct, and part of the uterine horn, the oviduct was positioned through changing the positions of the serrefine clamp and the hamster. The wall of the oviduct between the infundibulum and ampulla was dissected with micro-spring scissors. The tip of the capillary containing the embryos was inserted into the cut, and then pushed further towards the ampulla. The embryos were expelled into the ampulla with a minimum amount of collection medium. Eight vitrified-thawed embryos with normal morphology (with 2 even fully-expanded blastomeres) were transferred to two oviducts of a black female. After embryo transfer, the reproductive organs were replaced in the abdominal cavity, and the body wall incisions were sutured. Recipients were allowed to deliver and raise their own black pups as well as to foster golden ones.
Statistical analysis
The data from embryo vitrification and transfer were analyzed using arcsine transformation, followed by one-way or two-way analysis of variance (ANOVA), as appropriate. A P value of <0.05 for effects of factors (media, programs, developmental stages and vitrification) or interactions between factors (developmental stages and vitrification) was considered significant. When any of the factors had significant effects on the parameters, a post hoc procedure with least significant difference (LSD) tests was used for multiple and pairwise comparisons between groups. Means from differential staining were compared by the independent-samples t-test. All data were analyzed with a computer program, SPSS for Windows (Version 16.0; SPSS Inc, Chicago, IL, USA).
Results
In vitro development of two-cell embryos vitrified using different cryoprotectant solutions
Effect of the concentration of cryoprotectants on in vitro development of vitrified two-cell hamster embryos.

*Total number of embryos in each group derived from three replicates; within each replicate, 8–15 embryos were treated. †Means and SEMs were calculated arithmetically and analyzed by one-way analysis of variance (ANOVA). a,b,c,d,eWithin a column, values with no common letters are significantly different (P < 0.05). –: not applicable.
In vitro development of two-cell embryos vitrified using different cryopreservation programs
Effect of different vitrification programs on in vitro development of vitrified two-cell hamster embryos.
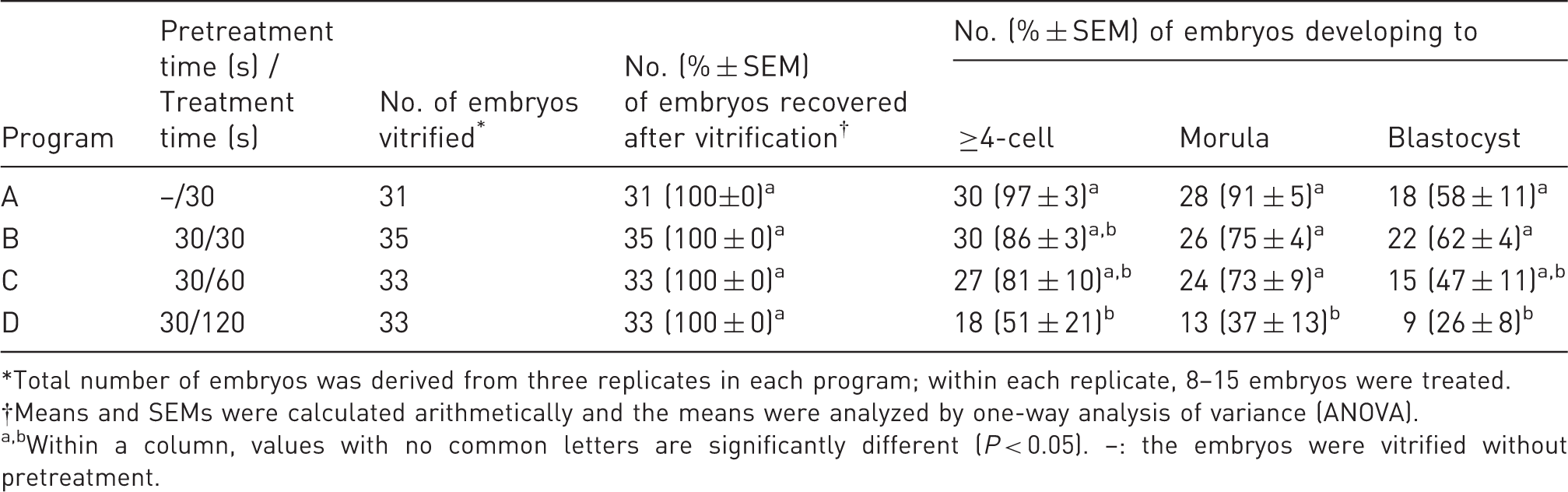
*Total number of embryos was derived from three replicates in each program; within each replicate, 8–15 embryos were treated. †Means and SEMs were calculated arithmetically and the means were analyzed by one-way analysis of variance (ANOVA). a,bWithin a column, values with no common letters are significantly different (P < 0.05). –: the embryos were vitrified without pretreatment.
In vitro development of embryos vitrified at different developmental stages
After establishing Program B to be the most effective vitrification program, we investigated the development potential of embryos vitrified at different developmental stages utilizing this program. Figure 1A shows the in vitro development of the embryos vitrified at different developmental stages. Pairwise comparisons showed no significant differences (P > 0.05) in blastocyst rates among vitrified two-cell (62%), four-cell (78%), eight-cell (80%), or morula (72%) embryos, but the blastocyst rates in all of these groups were significantly higher (P < 0.01) than that of the vitrified blastocysts (24%). Most dramatically, the in vitro development of vitrified PN embryos was arrested at the two-cell stage, even though 51% of PN control embryos developed to blastocysts. During the experiments, we also monitored the development of unvitrified control embryos isolated at the four-cell, eight-cell and morula stages. As shown in Figure 1A, embryos isolated at the four-cell, eight-cell and morula stages had high developmental ability with blastocyst rates of 91%, 92%, and 96%, respectively (with no significant differences among them); however, the blastocyst rates of embryos isolated at PN or the two-cell stage (51% or 79%, respectively) were significantly lower (P < 0.05) than embryos isolated at the four-cell, eight-cell and morula stages. When vitrified and unvitrified controls were compared at each of the corresponding developmental stages, no significant differences (P > 0.05) were observed between vitrified and control embryos at the two-cell, four-cell, or eight-cell stage, except for the embryos vitrified at morula or blastocyst stage (Figure 1A).
(A) In vitro development of hamster embryos at different developmental stages vitrified using EDFS30 media. The blastocyst rate was expressed as mean (%) + SEM, and the means were analyzed by two-way analysis of variance (ANOVA). PN, 2 C, 4 C, 8 C, M, and B: pronuclear, two-cell, four-cell, eight-cell, morulae, and blastocyst stage, respectively. a,bWithin each developmental stage, values with no common letters are significantly different (P < 0.05). A–DAmong vitrified or control groups, values with no common letters are significantly different (P < 0.05). (B) Differential staining of a hamster blastocyst derived from in vitro culture of vitrified/thawed two-cell embryos. (a) Inner cell mass (ICM) cells identified by their red fluorescence; (b) Total cells identified by their blue fluorescence; (c) A merged photograph shows ICM and trophectoderm (TE) cells. Scale bar equals 60 µm.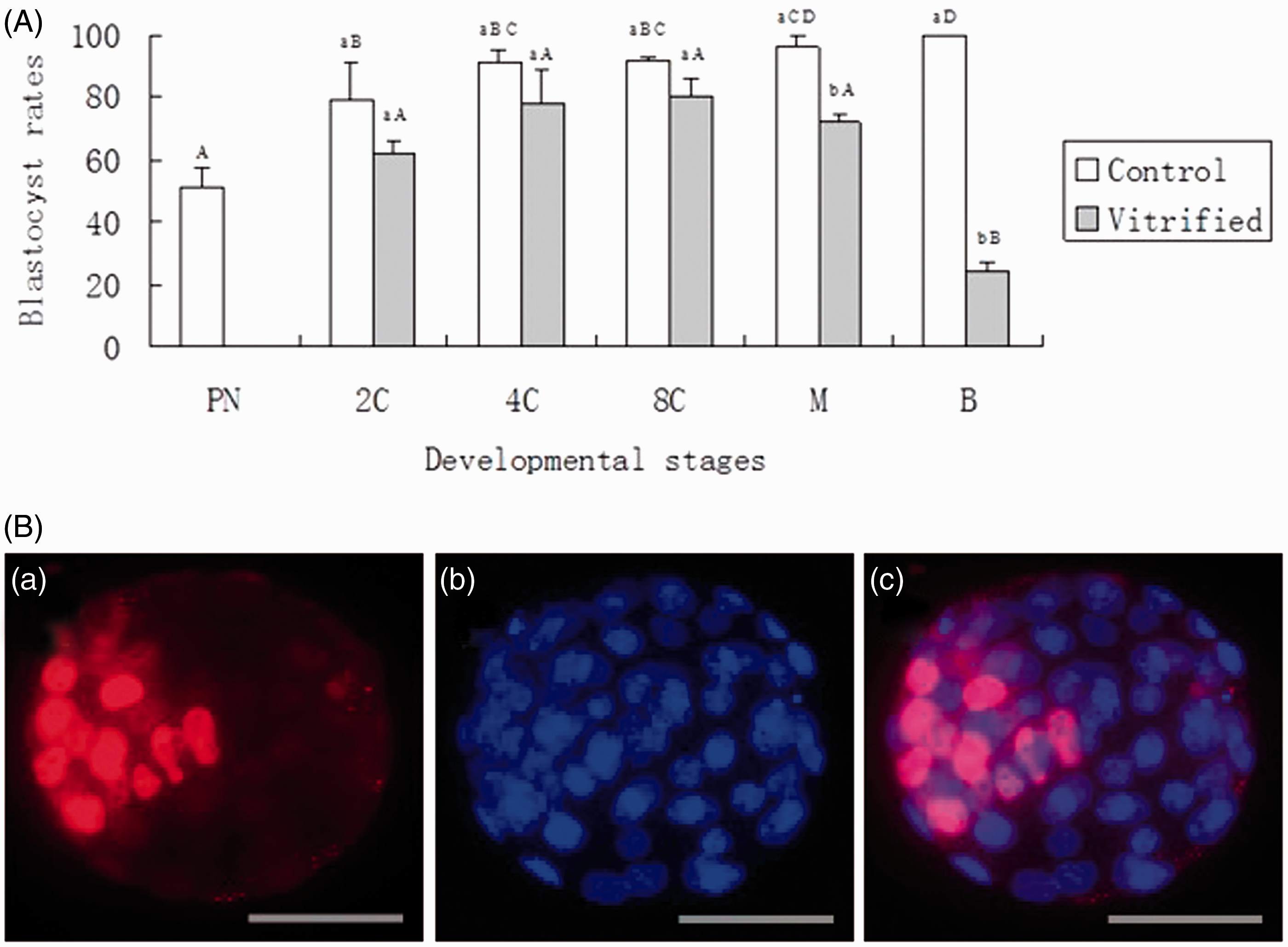
Differential staining of blastocysts derived from vitrified two-cell embryos
Differential staining of blastocysts derived from vitrified two-cell hamster embryos.

Values are means ± SEM where indicated, and the means were analyzed by the t-test.a,bWithin a column, values with no common letters are significantly different (P < 0.05). ICM: inner cell mass; TE: trophectoderm cells.
Embryo transfer
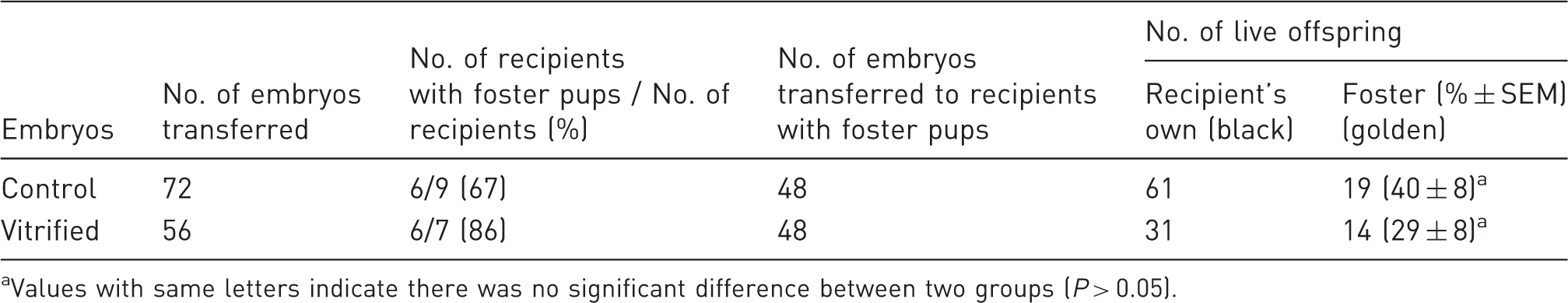
Four vitrified two-cell embryos that appeared morphologically normal after thawing were transferred to each oviduct of the recipients. Of the seven transfers performed, six (86%) resulted in pregnancies and the production of normal offspring. Figure 2 shows a surrogate mother with two fostered golden pups derived from vitrified two-cell embryos (along with six black pups derived from unmanipulated embryos). In total, 14 pups (29%) were born from transferring 48 vitrified embryos to recipients. As shown in Table 4, there was no significant difference (P > 0.05) in live birth rates between the vitrified embryos and the control embryos.
The black surrogate mother with her own six black pups and two foster golden pups derived from oviduct transfer of eight vitrified/thawed two-cell embryos. The pups were at the age of 10 days. In vivo development of two-cell hamster embryos vitrified by EDFS30 media. Values with same letters indicate there was no significant difference between two groups (P > 0.05).
Discussion
EG and DMSO have been widely used as permeating cryoprotective agents for embryo vitrification. 38 EG is less toxic to embryos and is a weak glass forming molecule, whereas DMSO is a better glass forming molecule. The two are therefore often combined in vitrification solutions. 39 It is quite often the case that solutions with 30% to 40% (or more) of permeating cryoprotectants are used for embryo vitrification. 40 In this study, we compared different concentrations of cryoprotectants in vitrifying hamster embryos. Our results indicated that EDFS30 was more suitable than EDFS35 or EDFS40 (Table 1) for the vitrification of two-cell hamster embryos. Cryoprotectant solution containing 15% EG and 15% DMSO has also been successfully used in the OPS vitrification of mouse 24 and goat embryos 41 and in cryoloop vitrification of human embryos, 42 but had not previously been used with hamster embryos. In the study reported by Lane et al., 19 cryoprotectant solution containing 20% of EG and 20% of DMSO was used in both the cryoloop and OPS vitrification experiments. Our success in using lower concentration of cryoprotectants to vitrify hamster embryos may have provided a much improved cryopreservation protocol for hamster embryos – the lower concentration of cryoprotectants means less toxicity to the embryos.
Besides the concentration of cryoprotectants, exposure time in vitrification solutions affects the survival and subsequent development of vitrified embryos. When exposure time was prolonged to 120 s, the in vitro developmental ability of hamster embryos was reduced (Table 2). This is likely due to toxic injury caused by EG and DMSO. 14 Therefore, to minimize toxic injury to embryos, we optimized the exposure time of embryos to cryoprotectant solutions. We found that 30 s and 60 s exposure times resulted in best blastocyst rates and that there was no difference (P > 0.05) in blastocyst rates between them. This finding indicates that exposing hamster embryos to cryoprotectant solutions for 30 s is enough to guarantee cryoprotectant permeation and to avoid intracellular ice formation; and this exposure time is comparable with the 25 s of exposure time reported in mouse 24 and goat embryos 41 during OPS vitrification. We also found that when 30 s of exposure time was used, there was no significant difference (P > 0.05) in blastocyst rates between embryos vitrified with and without pretreatment. This finding is unexpected in light of what has been observed in other species. For example, Zhou et al. 24 have demonstrated higher developmental potential of mouse earlier stage embryos vitrified by the two-step method than by the one-step method; Han et al. 43 have also reported that survival rates and in vitro development of vitrified rat two-cell embryos are improved by the two-step method. The difference in species (hamsters vs. mice or rats), components of vitrification solutions, and embryonic cultural conditions may account for such a discrepancy. Even though similar blastocyst rates were observed between the two methods in our study, pretreatment for a short time with lower concentration of cryoprotectants may give embryos more time for permeation and dehydration, which may be beneficial in providing effectiveness and stability to OPS vitrification. Therefore, we chose the two-step method as the preferred protocol for OPS vitrification of two-cell hamster embryos.
Comparing the embryos vitrified at different developmental stages (from two-cell to morula), no significant difference (P > 0.05) in blastocyst rates (62–80%) was observed. Therefore, the present protocol developed for two-cell embryos may also be used for vitrifying hamster embryos at four-cell, eight-cell, and morula stages. However, we found that vitrification with EDFS30 had a detrimental effect on PN and blastocyst embryos – no vitrified PN embryos developed to the blastocyst stage and a lower proportion (24%) of blastocyst stage embryos recovered after cryopreservation. A previous study has shown that the permeability of embryo cell membranes increases further as the developmental stage proceeds. 44 It is possible that the optimum condition developed for two-cell embryos may not achieve sufficient permeation for PN stage embryos because of the relatively lower surface area of PN embryos. Insufficient permeation and subsequent ice formation may account for lower developmental ability. For the lower recovery rate of vitrified blastocysts, we suspect that injures to the embryos caused by ice formation in the fluid-filled blastocoel during cryopreservation could be an underlying cause. 45 To prevent this, longer exposure and more permeation should be used for PN and blastocyst vitrification, but toxicity of cryoprotectants has to be taken into consideration.
The significant reduction of the total cell number and slight increase of the ratio of ICM to total cells (Table 3) indicate a delayed development of vitrified two-cell embryos. Reduction of blastomeres has been reported in vitrified mouse, 46 bovine, 47 and human 48 embryos. However, Lane et al. 19 observed no effect of vitrification on the total cell number in blastocysts derived from vitrified two-cell hamster embryos. One of the possible reasons for the discrepancy in results is that the blastocysts for cell counting were collected at earlier stages by Lane et al., since the total cell number (19.6 ± 1.4, OPS vitrification) 19 is much lower than that (48.7 ± 3.3, Table 3) in our study.
When vitrified two-cell embryos were transferred to recipients, 29% of them developed to live pups, which was comparable to fresh embryos (with a rate of 40%). The results of this study validate our successful establishment of a reliable OPS vitrification protocol to cryopreserve two-cell hamster embryos. Since there were no significant differences (P > 0.05) in blastocyst rates observed between fresh and vitrified embryos at the two-cell, four-cell, or eight-cell stages, we further posit that vitrified four-cell or eight-cell embryos are suitable for embryo transfer.
Our conclusion is that we have established an effective two-step OPS vitrification protocol for golden Syrian hamster embryos. This technological advancement provides a reliable method of cryopreserving hamster embryos and should greatly facilitate the development, propagation, distribution and application of genetically-engineered hamster lines.
Footnotes
Declaration of conflicting interests
Zhongde Wang is a co-founder of Auratus Bio, LLC, a biotechnology company specializing in creating genetically-modified animals for biomedical research and agricultural applications.
Funding
This work is supported by the Utah Science Technology and Research (USTAR) Initiative to ZW.