
Abstract
Poly(ADP-ribose) polymerase (PARP) is a highly abundant nuclear enzyme which metabolizes NAD, in response to DNA strand breakage, to produce chains of poly(ADP-ribose) attached to nuclear proteins. PARP activation has been implicated in ischemia/reperfusion injury, but its biological significance is not fully understood. We have modified an existing in situ method for detection of PARP activity by using an NAD analogue in which adenine is modified by an “etheno” (vinyl) bridge. Etheno-NAD serves as a PARP substrate in an initial enzymatic reaction; a specific antibody to ethenoadenosine is then used in an immunohistochemical reaction to detect the production of modified poly(ADP-ribose). The method produces strong and specific labeling of nuclei in which PARP has been activated, i.e., those in which DNA strand breaks have been produced, and the results can be analyzed by microscopy, flow cytometry, or colorimetry. The method is applicable to cultured cells in several formats and to frozen tissue sections. The particular characteristics of the new method may assist in future in situ studies of PARP activation.
Keywords
P
Extensive studies of the biological function and significance of PARP under normal and pathological conditions have suggested roles in many important biological processes, including differentiation, DNA replication, development, the response to DNA damage, apoptosis, gene expression, immunoglobulin heavy chain class switching, and regulation of chromatin structure and enzyme activity. However, more recent studies of two PARP “knockout” mouse models (de Murcia et al. 1997; Wang et al. 1995, 1997), as reviewed by Le Rhun et al. (1998), appear to show that PARP plays an important role in only a small number of processes. Perhaps the most important of these, from a clinical standpoint, is the response to ischemia/reperfusion of heart or brain. Global measures of injury (e.g., infarct volume) in animal models are substantially reduced with PARP inhibition by pharmacological or genetic means (Eliasson et al. 1997; Endres et al. 1997; Thiemermann et al. 1997). Nevertheless, more specific questions remain about PARP's role in this process (and possibly others that studies of PARP knockout mice may not have identified), e.g., whether all cells are involved equally, the time course of PARP activation, and whether PARP activation necessarily equates with ultimate death.
There are a number of means for detecting and measuring PARP activity, defined here as the synthesis of poly(ADP-ribose) (Shah et al. 1995). The most sensitive and quantitative of these are “aggregate” methods, which detect a signal generated by modified adenine in units of poly(ADP-ribose). For example, after incubation of cells with NAD radiolabeled in the adenine portion, PARP activity is proportional to the radioactivity incorporated into washed protein residues (Berger et al. 1978). However, when such methods are applied to the study of mixed cell populations, e.g., tissues, they do not indicate which cells are the source of the signal. An immunohistochemical staining method for the in situ detection of PARP activity, variously modified since its original description (Ikai et al. 1980), uses antibodies to unlabeled poly(ADP-ribose). This is more qualitative and less sensitive than other methods but gives information about PARP activity in individual cells.
Here we describe a modified in situ method to measure PARP activity, based on immunodetection of etheno-modified adenine incorporated into poly(ADP-ribose). Chemical modification of adenine, even within NAD, can be accomplished using chloroacetaldehyde to deposit an etheno (vinyl) bridge across the 1 and N6 positions (Barrio et al. 1972). Etheno-modified nucleotides, including ∊-NAD, may serve as substrates for many enzymes, including PARP in one isolated report (Farina et al. 1980). Etheno-modified nucleotides are intensely fluorescent, a property that has been exploited for sensitive, direct detection of nucleotide levels (Ramos-Salazar and Baines 1985; Tseng et al. 1994). It has also been utilized in a sensitive aggregate method to detect PARP activity. Isolation of unlabeled poly(ADP-ribose) from cells is followed by digestion into monomeric units, chemical conversion of their adenine into ∊-adenine, and quantitative measurement of fluorescence (Sims et al. 1980). However, no in situ method has been reported based on the fluorescence of ∊-adenine within poly(ADP-ribose). Despite the high quantum yield and large Stokes' shift of ∊-adenine (peak emission 420 nm), the wavelength for its excitation (peak 294 nm) is below the range of standard fluorescent microscopes containing glass (Barrio et al. 1972). Furthermore, it is unlikely that even an aggregate method to measure the fluorescence of ∊-adenine within poly(ADP-ribose) would be successful, because it has been shown that ∊-adenine fluorescence is highly quenched when it is part of a nucleic acid polymer, increasing with the proportion of adenine units that are etheno-modified (Lehrach and Scheit 1973; Steiner et al. 1973). Rather than fluorescence, we have used a primary antibody specific for ∊-adenosine to detect its presence within poly(ADP-ribose) in an immunohistochemical method to detect PARP activity in situ.
Materials and Methods
Reagents
1,N6-etheno-NAD (∊-NAD) was purchased from Sigma (St Louis, MO), as were all other reagents unless otherwise specified. Hybridoma supernatant of 1G4, a mouse IgG2aλ antibody specific for ∊-adenosine (Young and Santella 1988), was kindly provided by Dr. Regina Santella of Columbia University. A matched isotype antibody of unknown specificity was obtained from Sigma (clone HOPC-1).
Samples and Preparation
For stimulation of PARP activity, cultured cells were exposed to various DNA fragmenting agents while still in culture. N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) is a known strong stimulator of PARP activity (Alvarez-Gonzalez and Althaus 1989) and was routinely used as a positive control. MNNG dissolved in DMSO (100 mM) and stored frozen was added to cultures at a final concentration of 200 μM for a 30-min incubation before harvest. Irradiation of cell suspensions with UVB (312 nm) light was accomplished with a Stratalinker 1800 illuminator (Stratagene; La Jolla, CA) at 30 J/M2/sec, in a thin layer in Petri dishes at room temperature (RT). Before UVB exposure, some cells were grown for 20 hr in medium containing 10 μM 5-bromo-2′-deoxyuridine (BrdU).
An initial gentle fixation step, when employed, was chiefly performed with the crosslinking agent dimethylsuberimidate (DMS), freshly dissolved in ice-cold buffers at a concentration of 5.4-20 mg/ml, for 10-30 min. Buffers employed were Na2HPO4 (pKa 9.0) or sodium bicine (pH adjusted to 8.5, also containing 1.8 mM CaCl2), both at 100 mM. Other crosslinking fixatives used, all from Pierce (Rockford, IL), were DTBP (dimethyl 3,3′-dithiobispropionimidate), an analogue of DMS containing a cleavable disulfide bridge, DSP [dithiobis(succinimidylpropionate)], an N-hydroxysuccinimide diester also containing a cleavable disulfide bridge, and DTSSP [dithiobis(sulfosuccinimidylpropionate)], a cell-impermeant analogue of DSP. To reduce nonspecific binding of primary antibody in some applications, unreacted functional groups of crosslinking fixatives were inactivated after fixation by exposure to a buffer containing protein or glycine (50-100 mM).
Permeabilization was employed in all staining procedures, except with a transfection agent (see below), because NAD does not cross cell membranes. Permeabilization was achieved with cold ethanol, which also served as an initial fixative, or with hypotonic lysis or detergents, usually after fixation. Hypotonic lysis was accomplished as previously described (Grube et al. 1991), using a buffer of 10 mM Tris-HCl (pH 7.8), 4 mM MgCl2, 1 mM EDTA, and 30 mM β-mercaptoethanol.
Slices 750 μm thick were prepared from fresh whole rat brains with a tissue slicer (Stoelting; Wood Dale, IL) and were kept in artificial cerebrospinal fluid (aCSF) until fixation. To induce DNA fragmentation and stimulate PARP activity, some slices were treated with sodium nitroprusside (SNP, a source of nitric oxide) and pyrogallol (a source of superoxide anion), freshly dissolved in water and added to aCSF at final concentrations of 2 mM each, for 30 min at 37C. Slices were fixed with DMS (20 mg/ml) for 30 min and then frozen in Tissue-Tek/OCT (Miles; Elkhart, IN) for storage at −80C. For detection of PARP activity in situ, cryostat sections 5 μm thick were placed on glass slides, briefly incubated in hypotonic lysis buffer containing 0.2% Brij-58, and then taken to the ∊-NAD incorporation step below.
Incorporation of ∊-NAD (“PARP Reaction”)
After permeabilization, cells were incubated with ∊-NAD at a final concentration of 400 μM, which is the approximate nuclear concentration of NAD in vivo, under conditions previously described for the in situ incorporation of NAD into poly(ADP-ribose) (Grube et al. 1991). Cells in suspension were reacted at a concentration of 107/ml in a solution containing 53% (by volume) hypotonic lysis buffer, one-third 3 × suspension reaction buffer (100 mM Tris-HCl, pH 7.8, and 120 mM MgCl2), 2.5% ∊-NAD (from a 16-mm stock solution in water, stored at 4C), and the rest water. As a negative control, some reaction mixtures also contained 2.5% volume of the PARP inhibitor benzamide in DMSO, to a final concentration of 10 mM, with correspondingly less water. Cells or sections on glass slides were rinsed with 1 × slide reaction buffer (100 mM Tris-HCl, pH 8, 1 mM dithiothreitol, and 10 mM MgCl2) at RT, then incubated with a reaction mixture containing similar amounts of ∊-NAD with or without benzamide. Reactions were performed at 37C for 20 min, followed by fixation with 4% paraformaldehyde (PFA) or 10% formalin. To establish that the reaction product was poly(ADP-ribose) containing ∊-adenosine, in some instances slides were subsequently digested with snake venom phos-phodiesterase (from Crotalus adamanteus, type I) purchased from Worthington (Freehold, NJ), as described by Berger et al. (1978).
In limited numbers of experiments, two nonlethal approaches were used to introduce ∊-NAD into living cells, so that ∊-NAD incorporation occurred “in vivo.” Transfection of ∊-NAD was accomplished with Lipofectin (Gibco BRL/Life Technologies; Gaithersburg, MD), using protocols suggested by the manufacturer, at a final concentration of 0.5-1 mM ∊-NAD. In J774 cells grown on chamber slides, ∊-NAD was also introduced by reversible permeabilization with ATP (Steinberg and Silverstein 1989). Cells were overlayered with various media without additional Ca++ or Mg++ and the following reagents added (final concentrations): EDTA (5 mM), ATP (0.2 mM), and ∊-NAD (0.4 mM). Permeabilization was reversed by the addition of MgCl2 to 10 mM. With both approaches, subsequent steps included brief fixation with 95% ethanol, fixation in formalin or PFA (10 min), permeabilization with 0.1% Triton X-100, and immunostaining with antibody 1G4.
Immunodetection
For certain cell types, hydrogen peroxide (3% in water) was initially used for 5 min at RT to inactivate endogenous peroxidase. For cells not permeabilized with detergent or alcohol, slides were also incubated with PBS containing 0.1% Triton X-100 for 5 min at RT. Slides were blocked with normal goat serum (NGS, 5% in PBS) for 5 min at RT, then incubated with primary antibody 1G4, diluted 1:10 in PBS with 1% bovine serum albumin, for 30 min at RT in a humid chamber. For visible light microscopy, slides were then immunostained as previously described, using vats of biotinylated goat anti-mouse secondary antibody and avidin-horseradish peroxidase (both from Jackson Immunoresearch Laboratories; West Grove, PA), development with 3,3′-diaminobenzidine, intensification with CuCl2, and counterstaining with hematoxylin or Neutral Fast Red (Bindl and Warnke 1986). For fluorescence microscopy, slides were incubated with fluoroscein-conjugated goat anti-mouse secondary antibody (Cappel/Organon Teknika; Durham, NC).
Colorimetric Quantitation of In Situ Assay
U251 glioblastoma cells were seeded in 96-well plates at various densities, allowed to adhere, and incubated under control conditions or with MNNG for 30 min. After fixation and permeabilization with cold ethanol, the PARP reaction was performed as described above for adherent cells, using various concentrations of ∊-NAD, with or without 10 mM benzamide, in a volume of 50 μl/well. After paraformaldehyde fixation and NGS blocking, wells were incubated with immunodetection reagents as above, then developed with 3,3′,5,5′-tetramethylbenzidine; absorbance was read at 650 nm.
Flow Cytometry
After the PARP reaction and paraformaldehyde fixation, suspended cells were incubated for 5 min with cold IFA medium: 10 mM HEPES (pH 7.4), 150 mM NaCl, 4% bovine calf serum, 0.1% Triton X-100, and 0.1% azide. Cells were resuspended at 107 cells/ml in 70% IFA medium, 10% NGS, 10% normal human serum (type AB), and 10% 1G4 supernatant (final dilution 1:10) and incubated on ice for 1 h or overnight. After washing in IFA medium, cells were incubated with fluorescein-conjugated goat anti-mouse secondary antibody (Pharmingen; San Diego, CA), washed, fixed again with paraformaldehyde, and analyzed with a FACStar flow cytometer (Becton-Dickinson; Mountain View, CA).
Results
The method to detect PARP activity reported here uses ∊-NAD as a substrate and antibody detection techniques in a variety of formats. With all, the pattern of results was clearly consistent with the interpretation that ∊-NAD was utilized as a substrate by activated PARP in individual cells and incorporated into an analogue form of poly(ADP-ribose) containing ∊-adenosine. A strong positive result was consistently obtained in cultured cells incubated with MNNG, a known strong stimulator of PARP activity in vitro. As shown in Figure 1A, microscopic methods showed the reaction product to be exclusively nuclear, as expected for the localization of poly(ADP-ribose). Similar strong staining was also produced by UVB irradiation, but only in cells that had first been incubated with BrdU.
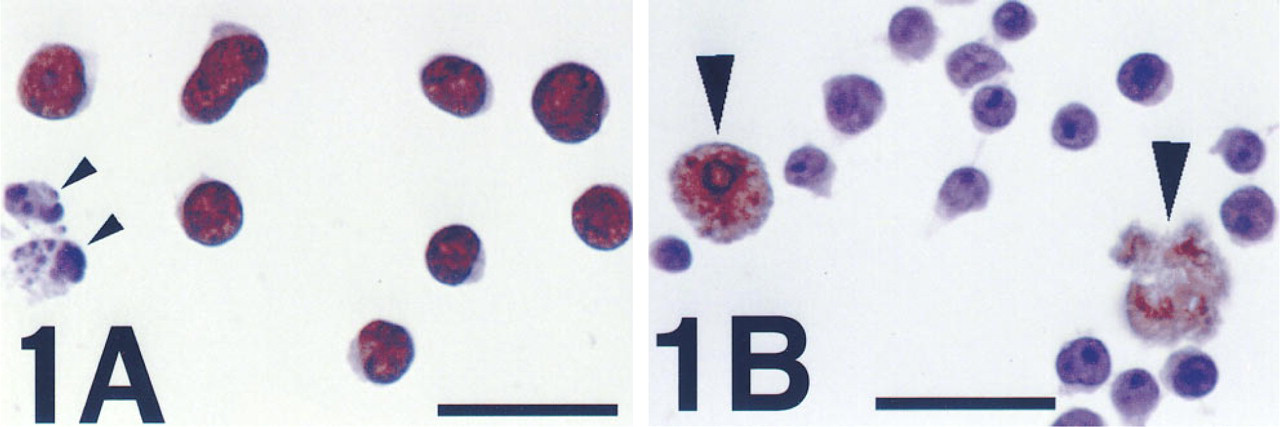
Jurkat cells immunostained for PARP activity, after cytocentrifugation and reaction with ∊-NAD.
It is known that UVB rapidly produces DNA strand breaks at sites of BrdU incorporation. The DNA-fragmenting effect of 312-nm light is increased 1000-fold by BrdU (Ben-Hur and Elkind 1972; Hutchinson 1973). To our knowledge, BrdU with UVB has not previously been used intentionally to stimulate PARP activity, although the modulation of its toxicity by PARP inhibitors in culture has been studied (Ben-Hur et al. 1985).
When the PARP reaction was performed in the presence of benzamide, a PARP inhibitor, all signal was abrogated, even with MNNG or BrdU/UVB. DMSO, the solvent used for adding benzamide to the PARP reaction, had no effect by itself at the concentration used (2.5%). Negative results were also obtained on omission of any of the necessary components, such as ∊-NAD or primary antibody, on addition of an excess of unlabeled NAD to the PARP reaction mixture, on replacing the primary antibody with a matched IgG2aλ isotype, and when cells were not permeabilized before (or during) the PARP reaction. After the PARP reaction, signal was also eliminated by incubation of MNNG-treated cells with snake venom phosphodiesterase, which cleaves poly(ADP-ribose) into free monomeric units. No staining was observed in control cells, except those whose morphology suggested that they had undergone spontaneous apoptosis in culture (Figure 1B). Although the staining in such cells was not exclusively nuclear, it was eliminated by incubation with benzamide. These results were a mirror image of those from MNNG-treated cultures, in which apoptotic cells showed no reaction product (Figure 1A). The latter observation is not surprising, because it is known that PARP is cleaved and inactivated during apoptosis by activated caspase-3 (Nicholson et al. 1995). Consistent with this, we found that the response of PARP activity to MNNG (or BrdU/UVB) is lost during apoptosis induced by several means (data not shown). It is unclear why PARP activation is detectable in cells that undergo spontaneous apoptosis. Using in situ detection, one group has found this in late stages (Donzelli et al. 1997), but another found it only in an early stage (Rosenthal et al. 1997).
Colorimetric measurement confirmed that staining results were indicative of the state of PARP activity, as defined by poly(ADP-ribose) synthesis. The aggregate signal produced by a fixed number of MNNG-treated U251 cells increased with the concentration of ∊-NAD but showed saturation typical of an enzymatic reaction (Figure 2A). Addition of benzamide to the PARP reaction reduced the optical density almost to background levels. Aggregate signal increased in linear proportion to the number of MNNG-treated U251 cells per well, above a certain number, again as expected for an enzymatic reaction (Figure 2B).
Flow cytometry also confirmed that the method could detect PARP activity in individual cells (Figure 3). After MNNG treatment and the complete PARP reaction, almost the entire population of cellular events was brightly fluorescent, shifted above the range of signal in MNNG-treated cells reacted in the presence of benzamide. In control cells, the complete PARP reaction produced a weak signal, as shown by the effect of adding benzamide to the PARP reaction mixture. Although qualitative microscopic evaluation of control cells found no definite staining (except in rare apoptotic cells), this weak signal with flow cytometry was consistent with the weak colorimetric signal produced by control U251 cells (Figure 2B). Conventional aggregate methods to measure PARP activity, using sensitive fluorimetric or radiometric methods to detect poly(ADP-ribose), also consistently show a weak but specific signal in control cells (Berger et al. 1978; Alvarez-Gonzalez and Althaus 1989).
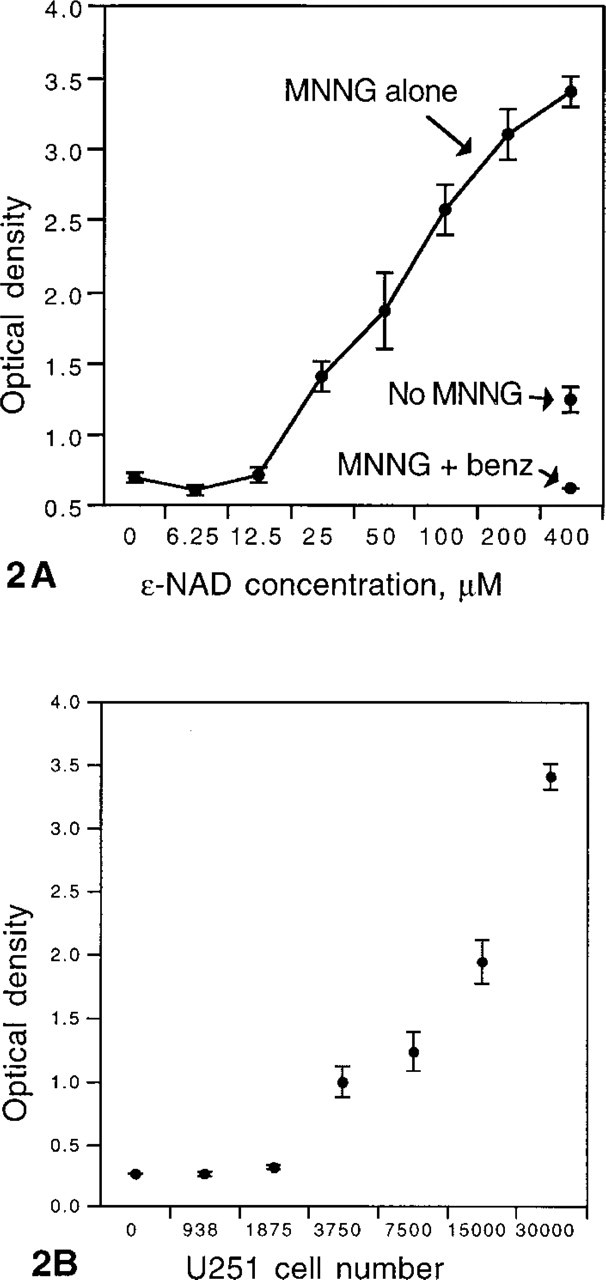
Colorimetric measurement of PARP activity in U251 cells. (A) The aggregate signal produced by a fixed number of MNNG-treated U251 cells (MNNG alone) increases with ∊-NAD concentration over most of its range. Although the increase in signal (optical density) appears linear, the ∊-NAD concentration is displayed in a logarithmic format and the data show saturation of PARP at higher ∊-NAD concentrations. At maximal ∊-NAD concentration (400 μM), there is some activity in control cultures (No MNNG). Even after MNNG treatment, addition of benzamide during the PARP reaction reduces the optical density nearly to blank levels (MNNG + benz). Values represent the mean of triplicate cultures (3 × 104 cells seeded per well). Error bars show the SEM.
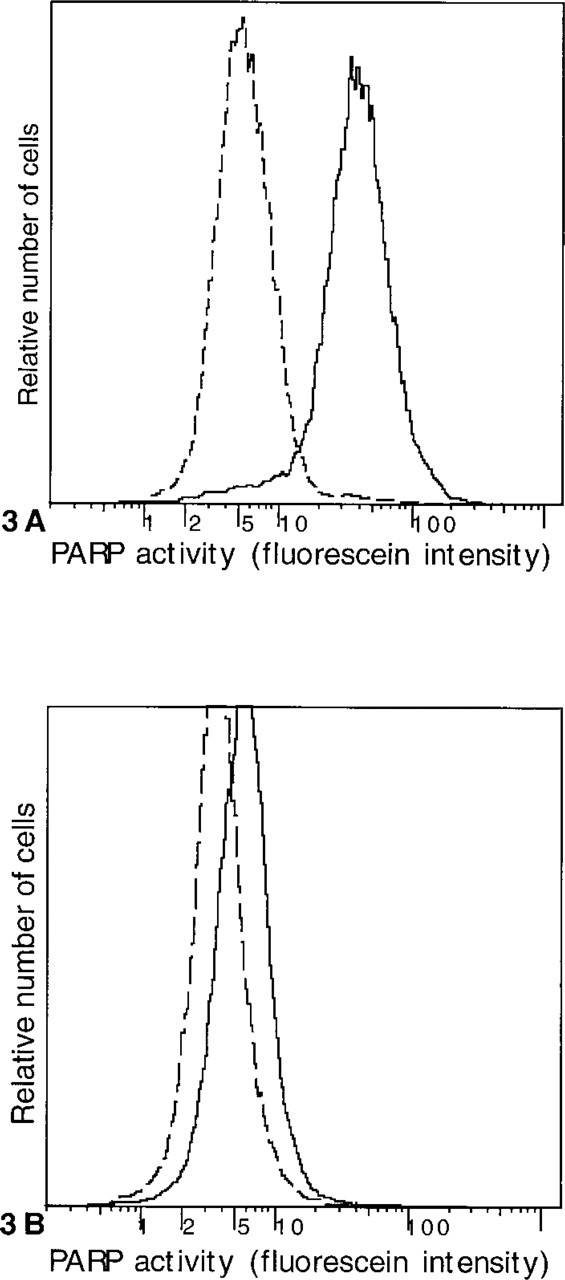
Flow cytometric detection of PARP activity in Jurkat cells. Cells were permeabilized by hypotonic lysis, reacted with ∊-NAD in the absence (solid line) or presence (dashed line) of benzamide, and immunostained with a primary antibody to ∊-adenosine and a fluorescein-conjugated secondary antibody. (A) MNNG produces a strong signal in the entire population of cells, which is abolished by benzamide.
Similar results were obtained in a wide variety of adherent and nonadherent cell types from both human and rodent sources, although not all were tested in all formats. Neoplastic cell lines examined included lymphoid (Jurkat and REH), myeloid (HL-60 and NB4), macrophage (J774), glial (U251), neural (PC-12 and SY5Y), squamous (SKOV-3), and melanoma (K1735). Positive results were obtained after induced differentiation using HL-60, NB4, and PC-12 cells, although this required stimulation by MNNG. PARP activity was also detected in non-neoplastic cells, i.e., in monolayer cultures of primary fetal rat brain (pure glial and mixed glial/neuronal).
As implied by its application to different cell types in various formats, the method was quite robust. In some cell types, PARP activity was successfully detected with many combinations of fixation and permeabilization steps, but other cell types had more specific requirements. Initial fixation was not absolutely required but was valuable according to the cell type and format involved. For microscopic methods, fixation was primarily important in the preservation of morphology. However, the effect of fixation on morphology varied considerably among cell types and was less if cells were immobilized on glass at an early point. DMS and related crosslinking fixatives were found to be the best overall, preserving PARP activity in all cell types. For some adherent cell types, fixation was important in preserving desired attachment to chamber slides, especially against subsequent permeabilization. Ethanol was found to be the best fixative in this respect, but it was also found to destroy PARP activity in certain cell types (HL-60). In PC-12 cells, no fixative was found to prevent detachment and preserve PARP activity before or after differentiation by nerve growth factor, but cells could still be stained after detachment, cytocentrifugation, and DMS fixation. In addition to ethanol and hypotonic buffer, various detergents were successful at permeabilization and subsequent PARP activity detection in one or more cell types. Those used were Triton X-100 (0.1-0.5%), Brij-58 (0.2%), digitonin (0.01%), lysolecithin (0.25 mg/ml), and saponin (0.1-0.5%).
With transfection or ATP permeabilization, the incorporation of ∊-NAD into poly(ADP-ribose), i.e., the PARP reaction, could be studied while cells were still in culture. With variation of the time of application of the stimulus to PARP activity, these approaches provided additional information about the metabolism of ∊-NAD, i.e., its duration as a substrate, its competition with native NAD, and the stability of the ∊-adenosine-labeled poly(ADP-ribose) in living cells. Furthermore, these methods also had the advantage that analysis was quite simple. At a given point, cells were simply harvested from culture, fixed, and immunostained with the antibody to ∊-adenosine. With Lipofectin, a cationic surfactant most commonly used for liposomal transfection of nucleic acid, we detected very strong nuclear labeling in a subset of HL-60 (Figure 4A) or U251 cells (Figure 4B) subsequently treated with MNNG. There was no staining of control cells or of cells cultured with benzamide during treatment with MNNG. The low frequency of staining was presumably due to low efficiency of ∊-NAD introduction. Similar “metabolic labeling” of cultured cells with nonradioactive nucleotide analogues by liposomal transfection has previously been described, e.g., RNA synthesis was detected in situ after introduction of bromo-UTP (Haukenes et al. 1997). There was additional evidence to support the assumption that, with this method, the metabolism of ∊-NAD by PARP (and perhaps other routes) takes place in a near-physiological manner. Subsequent culture showed no toxicity due to ∊-NAD transfection alone, and after 24 h this approach could not detect MNNG-stimulated PARP activity.
J774, a mouse macrophage cell line, can be permeabilized by ATP in a manner that is reversible and is related to the relative concentrations of ATP4- and divalent cations (Ca++ and Mg++). Dyes up to 831 Da, exceeding the molecular weight of ∊-NAD (687.5 Da), can be rapidly introduced (Steinberg et al. 1987). By this means we were able to achieve specific nuclear labeling in cultures stimulated with MNNG or BrdU/UVB. Staining was not as intense as that achieved with Lipofectin in HL-60 cells but was present in almost all cells (Figure 5A). Staining was abrogated by omission of the PARP stimulus, the substrate (∊-NAD), or the permeabilizing agent (ATP), and by the addition of benzamide. Staining was most intense when the medium used for ATP permeabilization was a simple salt solution, either one found to be most effective for the introduction of dye into J774 cells (135 mM NaCl, 5 mM KCl, and 25 mM Tris, pH 8.0) (Steinberg et al. 1987) or one similarly used to introduce small molecules into transformed 3T3 fibroblasts (50 mM NaCl, 50 μM CaCl2, and 100 mM Tris, pH 8.2) (Rozengurt and Heppel 1975). Staining was less intense when the permeabilization medium was Dulbecco's modified essential medium (DMEM), which contains Ca++ or Mg++, and least when the critical agents (EDTA, ATP, and ∊-NAD) were simply added to the medium in which the cells were growing (DMEM with 10% fetal calf serum and antibiotics). The intensity of staining was also related to the time of addition of MNNG, being greatest when MNNG was added 30-60 min before ATP permeabilization and ∊-NAD introduction. When the latter processes were stopped by adding MgCl2 before MNNG, staining was barely detectable, even for a comparable duration of MNNG exposure. This result may be evidence that ∊-NAD incorporation into poly(ADP-ribose) is reduced by competition with native NAD in cells. The dose of MNNG used was one that reduces NAD levels to <10% of normal in 30 min (unpublished results, and Berger 1985). However, an alternative explanation would be that introduced ∊-NAD is metabolized during the period before MNNG treatment. Normal mouse macrophages possess high levels of an NADase ectoenzyme (Artman and Seeley 1979), similar to an NADase in calf spleen cells, which can metabolize ∊-NAD (Muller et al. 1983). Furthermore, we found that staining was reduced when higher levels of ATP were used for permeabilization, perhaps because ATP is a substrate for NAD synthesis. In some experiments, cells were initially exposed to MNNG, permeabilized with EDTA/ATP/∊-NAD, and then left in culture after addition of MgCl2 (to stop further ATP-mediated introduction of ∊-NAD) and benzamide (to prevent further ∊-NAD incorporation). Within 30 min, staining was considerably reduced (Figure 5B), which we interpret as evidence that e-adenosine-labeled poly(ADP-ribose) is susceptible to degradation in vivo. Native poly(ADP-ribose) is rapidly degraded by a specific enzyme, poly(ADP-ribose) glycohydrolase, which opposes PARP and is activated by the same conditions that activate PARP (Alvarez-Gonzalez and Althaus 1989).
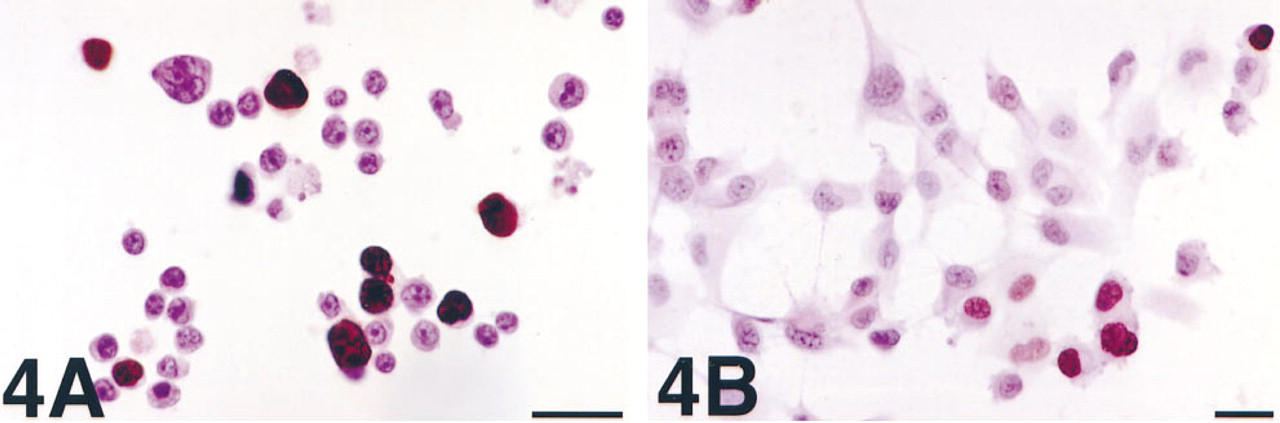
PARP activity detection in HL-60 cells
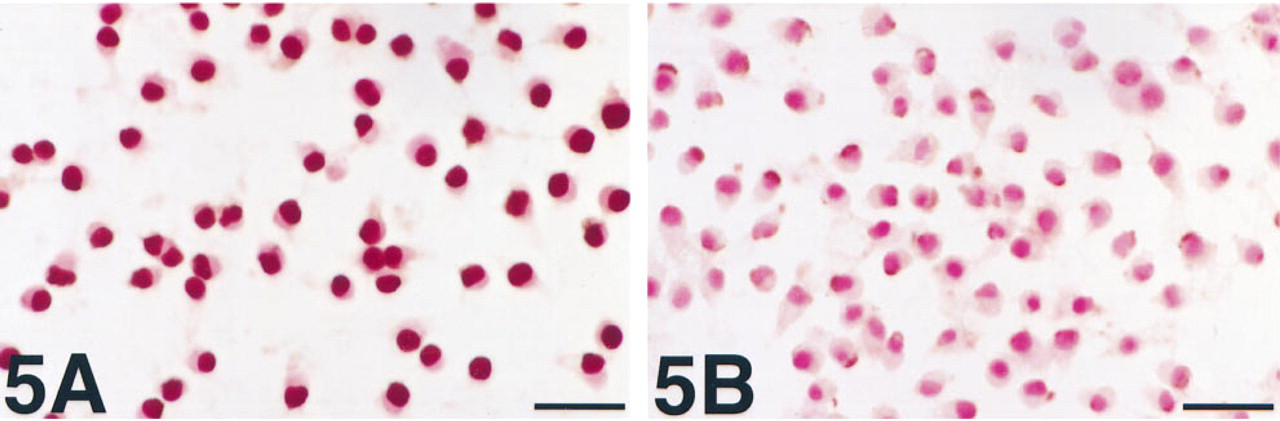
PARP activity detection in J774 cells in which ∊-NAD has been introduced with ATP.
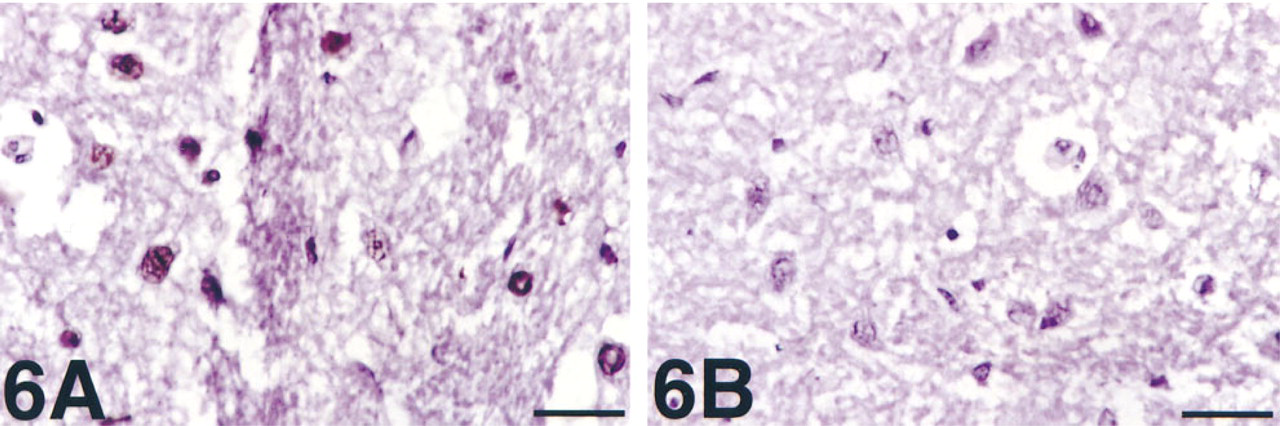
PARP activity detection in frozen sections of rat brain slices incubated with SNP and pyrogallol. Strong, exclusively nuclear staining is seen.
The method was also able to detect PARP activity in frozen sections prepared from slices of rat brain. Slices that had been incubated with SNP and pyrogallol between harvesting and fixation were found to have strong nuclear reactivity (Figure 6A). SNP and pyrogallol together generate peroxynitrite, a DNA-fragmenting agent and PARP stimulator believed to mediate some of the injury of ischemia/reperfusion (Szabó and Salzman 1995; Eliasson et al. 1997). There was no reactivity in non-nuclear areas, in control slices incubated without SNP and pyrogallol, when the primary antibody was omitted, or when benzamide was added to the PARP reaction mixture (Figure 6B).
Discussion
In this report we describe a novel method for the in situ detection of PARP activity, defined as the capacity for poly(ADP-ribose) synthesis. The method is based on two essential phenomena: (a) the ability of PARP to utilize ∊-NAD, an analogue of its normal substrate (NAD), and (b) the ability of a specific antibody to recognize the ∊-adenosine moiety after incorporation into an analogue form of poly(ADP-ribose). The method is derived from earlier in situ methods to detect PARP activity, which use antibodies to poly(ADP-ribose) and immunofluorescent detection. The original method used rabbit antibody in rat cells or frozen sections after ethanol fixation alone (Ikai et al. 1980). This is a simple approach, which in theory reflects the native level of poly(ADP-ribose). However, it was also found that stronger staining was obtained after incubation with an excess of NAD, and most subsequent studies have used permeabilization and incubation with NAD. Further developments of this method have continued, including the use of a monoclonal antibody (clone 10H) to poly(ADP-ribose) (Kawamitsu et al. 1984), additional permeabilizing agents and fixatives (Grube et al. 1991), detection by peroxidase immunohistochemistry (Küpper et al. 1996), “tricolor” combination with fluorescent markers for nuclear changes and DNA end-labeling in apoptosis (Negri et al. 1997), and application to frozen sections of brain after ischemia/reperfusion (Eliasson et al. 1997).
Although the present method similarly detects PARP activity on the basis of poly(ADP-ribose) production, the different substrate and antibody used may provide greater sensitivity. The epitope of unlabeled poly(ADP-ribose) bound by antibody 10H, most commonly used in previous in situ studies, appears to be a linear region of several monomeric units (Kawamitsu et al. 1984). This antibody might therefore have suboptimal sensitivity to small amounts of poly(ADP-ribose) or to branched regions of poly(ADP-ribose). The relative abundance of the latter is increased after MNNG treatment, and they are degraded more slowly than are linear regions (Malanga and Althaus 1994). In contrast, ELISA studies have shown that antibody 1G4 recognizes pure e-adenosine with exquisite sensitivity and specificity (Young and Santella 1988). Therefore, aside from possible steric factors, 1G4 should be able to bind to single ∊-adenosine moities incorporated into poly(ADP-ribose), independent of polymer configuration.
The different substrate and antibody used in the present method may also yield greater specificity in the detection of poly(ADP-ribose) synthetic capacity, i.e., PARP activity, at a particular point or period of interest. Even though most linear poly(ADP-ribose) is rapidly degraded by poly(ADP-ribose) glycohydrolase after MNNG exposure, the rate of degradation declines dramatically as chains become shorter, and there is a baseline of short polymers attached to PARP and other nuclear proteins (Alvarez-Gonzalez and Althaus 1989; Malanga and Althaus 1994). Therefore, methods using antibodies to poly(ADP-ribose), with or without permeabilization and incubation with unlabeled NAD, can be confounded by polymer synthesized before the point or period of interest. The present method is not subject to this potential problem because ∊-adenine is not a normal cell constituent. Although ∊-adenine can be produced by vinyl chloride exposure and ethenoadenine binding intracellular proteins have been reported (Dosanjh et al. 1994), ∊-NAD serves as a novel substrate for assay of PARP activity. (We found no evidence that such proteins interfered with the specificity of our method.) Accordingly, antibody 1G4 should not bind to preexisting poly(ADP-ribose) or to other cell constituents, regardless of whether or not ∊-adenine is incorporated into newly synthesized poly(ADP-ribose).
These attributes also allow ∊-NAD to serve as a nonradioactive label in various studies of PARP activity and related metabolism in living cells, although special means to introduce ∊-NAD are required. Compared to transfection, ATP permeabilization is simple and inexpensive, and in our experiments produced a much higher proportion of labeled cells. However, it can be applied only to a limited number of cell types. Because radiolabeled adenine (Alvarez-Gonzalez and Jacobson 1987; Aboul-Ela et al. 1988) or adenosine (Tanuma and Johnson 1983; Malanga and Althaus 1994; Althaus et al. 1995) has previously been used to detect PARP activity in living cells after simple addition to cultures and overnight incubation, we tried a similar approach using ∊-adenosine. However, no positive results could be obtained. In living cells, radiolabeled adenine and adenosine are converted to ATP, which can then be incorporated into NAD by the enzyme NAD pyrophosphorylase (NMN adenylyltransferase; EC 2.7.7.1). This enzyme appears to channel NAD to PARP in the nucleus, such that radioactive ATP is rapidly incorporated into poly(ADP-ribose) (Uhr and Smulson 1982). We applied ∊-ATP directly to permeabilized cells in vitro or in vivo but, again, no positive results were produced. This suggests that e-ATP cannot serve as a substrate for NAD pyrophosphorylase. Other nucleotides containing ∊-adenine, including ∊-adenosine (Dutta et al. 1980) and ∊-NAD (Barrio et al. 1972), are known to vary considerably among enzymes as to their suitability as substrates or cofactors (Leonard 1984). Indeed, our results confirm that ∊-NAD can serve as a substrate for the poly(ADP-ribose) synthetic function of PARP and suggest that ∊-adenine in poly(ADP-ribose) does not prevent its enzymatic degradation, but ∊-NAD cannot serve as a substrate for the NAD glycohydrolase function of PARP in human HL-60 cells (Kirsten et al. 1991).
Even though this method is an assay of enzyme activity, we found that initial fixation was a critical variable for most cell types. DMS is a homobifunctional crosslinking agent which forms imido diesters with free amino groups, principally at lysine residues. DMS has previously been shown to provide excellent fixation of ultrastructure in tissues, with remarkable preservation of enzyme activity by cytochemical staining (Hassell and Hand 1974; Hand and Hassell 1976). Furthermore, DMS and related fixatives have even been found to enhance the molar activity of certain enzymes in purified preparations (Hartman and Wold 1967; Wang and Moore 1977). The molar activity of purified PARP in vitro is increased by glutaraldehyde, owing to the sensitivity of PARP's molar activity to its concentration (Bauer et al. 1990). Most evidence suggests that PARP functions in vivo as a homodimer (Alvarez-Gonzalez and Mendoza-Alvarez 1995), and crosslinking fixatives appear to preserve activity by preserving a dimeric state of self-association. However, biochemical studies do not always predict performance in staining methods. We found that concentrations of glutaraldehyde used for electron microscopy destroyed all staining with our method, even though paraformaldehyde left some PARP activity. Further-more, DMS binds principally to lysine residues, but mutation analysis has shown that a lysine residue is critical to PARP's catalytic activity (Simonin et al. 1990). Whether fixation inactivates PARP may be a function of whether DNA binding and/or NAD catalytic sites are occupied, because this has been found for other enzymes. We found that fixation was also important to adherence in the staining of monolayers, especially during permeabilization; crosslinking fixatives preserve cytoskeletal elements against permeabilization (Safiejko-Mroczka and Bell 1996).
As an in situ technique, this method can provide information about PARP activity in individual cells, at the same time allowing correlation with other features of the cells, e.g., morphology in microscopic formats and other parameters in flow cytometry. The basic method uses simple solutions, standard immunostaining reagents, and a commercially available nucleotide analogue (∊-NAD) which is relatively inexpensive. [A recent report suggests that biotinylated or digoxigenin-labeled NAD can be used in a similar approach (Zhang 1997).] The versatility of the method has been shown in a variety of formats. Some of these (flow cytometry and colorimetry) are novel for an in situ method to detect PARP activity, but the most exciting may be frozen tissue sections. The method requires special preparation of fresh samples, but this is true for most techniques that detect enzyme activity. This method may assume a place alongside other techniques to measure PARP activity and may help in elucidating the biological role of an enzyme with such high abundance and catalytic potential.
Footnotes
Acknowledgments
RED was supported by an Institutional Research Grant from the American Cancer Society. JCB was supported by the Pfeiffer Foundation Minority Summer Research Program and the Stanford Medical Scholars Program. JCH was supported by Dr Richard Zare and by an undergraduate research award from the Camille and Henry Dreyfus Foundation. Q-ATL and PDK were supported by NIH grant CA 42509 to Leonard A. Herzenberg.
We gratefully acknowledge the support of the following individuals: for cell lines, Michael Hsaio, Philipp Kahle, Eric Shooter, Esther Chang, Kevin Smith, Yacop Jacobs, Louie Naumovski, and Doug Ross; for primary cultures, Robert Sapolsky, Rona Giffard, and Michelle Emond; for brain samples, Brie Linkenhofer, Eric Schaible, and Daniel Madison; for preparation of frozen sections and assistance with immunostaining, Eva Pfendt; for expert secretarial and photographic assistance, Eileen Maisen and Phil Verzola; and for sponsorship and critical reading of the manuscript, Roger Warnke, Leonard Herzenberg, and Michael Cleary.