
Abstract
We describe a new freeze-fracture cytochemical technique consisting of combined immunocytochemistry and enzyme cytochemistry. This technique reveals the relationship between molecules in biological membranes by double labeling with two different cytochemical markers (i.e., immunogold probes and cerium). In this method, antigens were detected with specific primary antibodies and appropriate secondary immunoprobes. Subsequently, alkaline phosphatase activity was detected with cerium as the capture agent on the same replicas. Octyl-glucoside (OG) digestion before the cytochemical reactions was crucial to the success of this combined method. OG is an efficient detergent and OG digestion can preserve both immunocytochemical antigenicity and enzyme activity on replicas. As an initial examination, we applied this technique to the study of glycosyl-phosphatidyl-inositol-anchored proteins and adhesion molecules in human neutrophils. The method described here should serve as a unique additional approach for the study of topology and dynamics of molecules in biomembranes.
Keywords
F
We introduce here octyl-glucoside (OG) digestion for this new double labeling method. Our results show that immunocytochemistry and enzyme cytochemistry can be combined to demonstrate the localization of two different proteins on the same replicas. The method described here should serve as a unique additional approach for the study of topology and dynamics of many molecules in biological membranes.
Materials and Methods
Reagents
The following chemicals were purchased from Sigma (St Louis, MO): Crium chloride, dextran, dimethyl sulfoxide (DMSO), β-glycerophosphate, Hank's balanced salt solution, Histopaque 1083, levamisole, Tricine, and N-tris methyl-3-aminopropanesulfonic acid (TAPS). Glutaraldehyde (25% aqueous), maleic acid, 1-O-n-octyl-β-
Mouse monoclonal anti-human leukocyte antigen (HLA) Class I (clone W6/32) and mouse monoclonal anti-human CD16 (Fc γ-receptor III) (clone DJ130c) were obtained from Dako (Glostrup, Denmark). Mouse monoclonal anti-human CD62L (L-selectin) (clone FMC46) was from Novocastra (Newcastle upon Tyne, UK). Mouse monoclonal anti-human CD16 (clone gran 1) was kindly provided by Dr. Clark L. Anderson (Ohio State University). Ten-nm colloidal gold was prepared by the tannic acid/citrate method and then conjugated with affinity-purified goat anti-mouse IgG (Cappel; Durham, NC) (Slot and Geuze 1985). All immunological reagents were handled in accordance with the manufacturers' recommendations and were used before the expiration date for each product.
Cell Isolation
Whole human blood was collected from healthy adult men after obtaining informed consent. Neutrophils were purified from whole blood in the unstimulated state, as described previously (Takizawa and Robinson 1993).
Cytochemistry on Replicas
Unstimulated human neutrophils were rapidly frozen and fractured as reported previously (Takizawa and Saito 1997b). Briefly, the neutrophils were quickly frozen in liquid nitrogen slush. The frozen cells were then freeze-fractured at −130C and subsequently replicated by Pt/C evaporation in a freeze-fracture apparatus (FD-2A; Eiko, Mito, Japan). The replicas were floated from the cells in PBS, rinsed in PBS two times, and then transferred to 4 ml of 0.6–60 mM OG-PBS solution. OG digestion was carried out for 1 hr at 4C.
After OG digestion, the replicas were rinsed with three changes of PBS and then incubated with the following antibodies for 60 min at room temperature (RT): anti-HLA Class I (33.0 μg/ml), anti-CD16 (clone: gran 1, 5 μg/ml; DJ130c, 2.7–5.4 μg/ml), anti-CD62L (diluted 1:10; this dilution was of the material supplied by the manufacturer). The replicas were then rinsed in PBS three times over 15 min. Detection of the primary antibody binding sites was achieved with a 60-min incubation in goat anti-mouse IgG conjugated with 10-nm gold. All antibody solutions were diluted with PBS containing 10% normal goat serum. The replicas were rinsed three times in PBS and then transferred to 0.1 M cacodylate buffer (pH 7.4) containing 5% sucrose.
Incubation for the detection of alkaline phosphatase (AL-Pase) activity was subsequently performed for 60 min at 37C with constant agitation. This medium contained 50 mM Tricine, 100 mM TAPS, 2 mM CeCl3, 5 mM β-glycerophosphate, 5 mM MgSO4, and 5% sucrose, a modification of the medium of Kobayashi and Robinson (1991). The reaction medium was pH 9.3. Then the replicas were rinsed in cacodylate buffer.
After the cytochemical reactions, the replicas were postdigested in PBS containing 20 mM OG, overnight at RT. The samples were then fixed in 2% glutaraldehyde in cacodylate buffer for 10 min at RT, washed with distilled water, collected on Formvar-coated copper grids, and examined with a Hitachi H-7000 electron microscope operated at 100 kV. During the entire procedure the replicas were floated on the various liquids employed.
Control immunocytochemical and enzyme cytochemical incubations consisted of omission of the primary antibodies from the immunoreaction mixture and inclusion of the alkaline phosphatase inhibitor levamisole (2.5 mM) in the complete enzyme reaction mixture, respectively.
In parallel experiments, we tested the possibility of using SDS, as reported by Fujimoto (1995), for digesting replicas for immunocytochemical labeling. The replicas were digested in 2.5% SDS solution for 1 hr at RT before the cytochemical reaction.
Morphometric Analysis of Immunogold in Human Neutrophils
The labeling density of 10-nm colloidal gold-IgG particles indicating the localization of HLA Class I was determined. Negatives of electron micrographs of the replicas were printed at the same magnification (X 63,000). The area of the exoplasmic halves (E-faces) of plasma membranes was measured and the number of individual immunogold particles with the E-faces was counted on the micrographs.
Results
The replica digestion before the cytochemical reactions was crucial for this new method. Experiments were carried out with various concentrations of OG in the replica digestion solution with the replicas. For immunocytochemical fracture-labeling, OG not only efficiently digested unfractured cell components but also adequately retained immunocytochemical antigenicity on the split membranes stabilized with Pt/C. OG at 0.6–60 mM gave good results for the antigens tested. For example, HLA Class I was detected primarily on freeze-fractured plasma membranes in resting human neutrophils (Figure 1A). For enzyme cytochemical fracture-labeling, OG was able to preserve enzyme activity on the replicas. ALPase activity was demonstrated in association with the freeze-fractured membranes of small intracellular granules (Figure 1B). Best results were found when OG concentrations of 6–20 mM were included in the digestion medium. Higher concentrations of OG (more than 20 mM) led to failure in detection of the reaction product for ALPase activity (Figure 2). In addition, lower concentrations of OG (less than 6 mM) led to poorer dissolution of unfractured cell components attached to the replicas. Such was the case even though intracellular enzyme cytochemical staining for ALPase was detectable. Optimal conditions for the use of OG in this combined cytochemical approach were found when the replicas were digested in 6–20 mM OG-PBS solution for 1 hr at 4C. In these experiments the detection of enzyme activity on replicas was more sensitive to the OG digestion than was detection of antigens (Figure 2). After SDS digestion, it was possible to detect antigens on replicas. However, the enzyme cytochemical results obtained in this case were not acceptable because ALPase activity was not detected (Figure 1C).
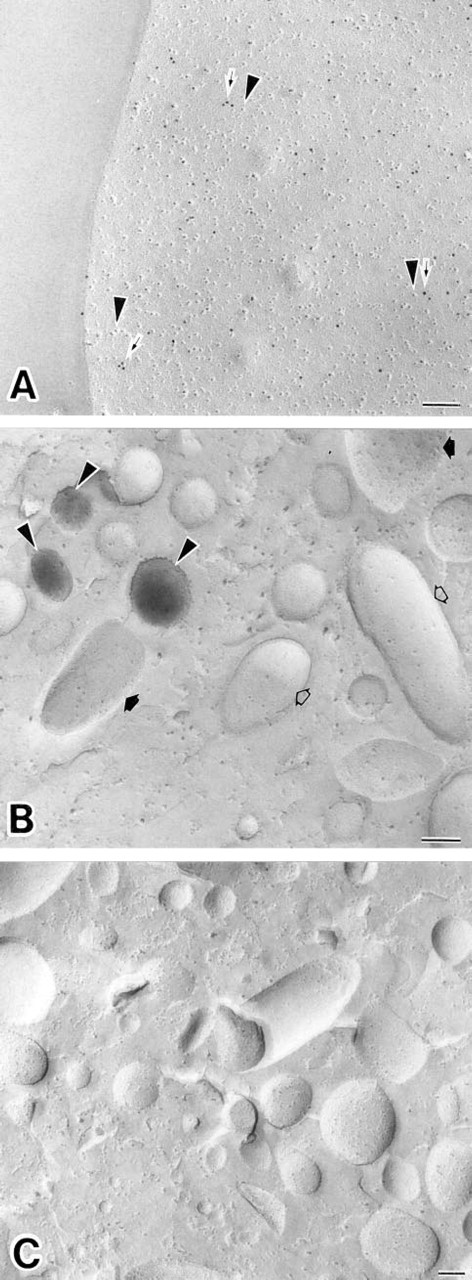
(
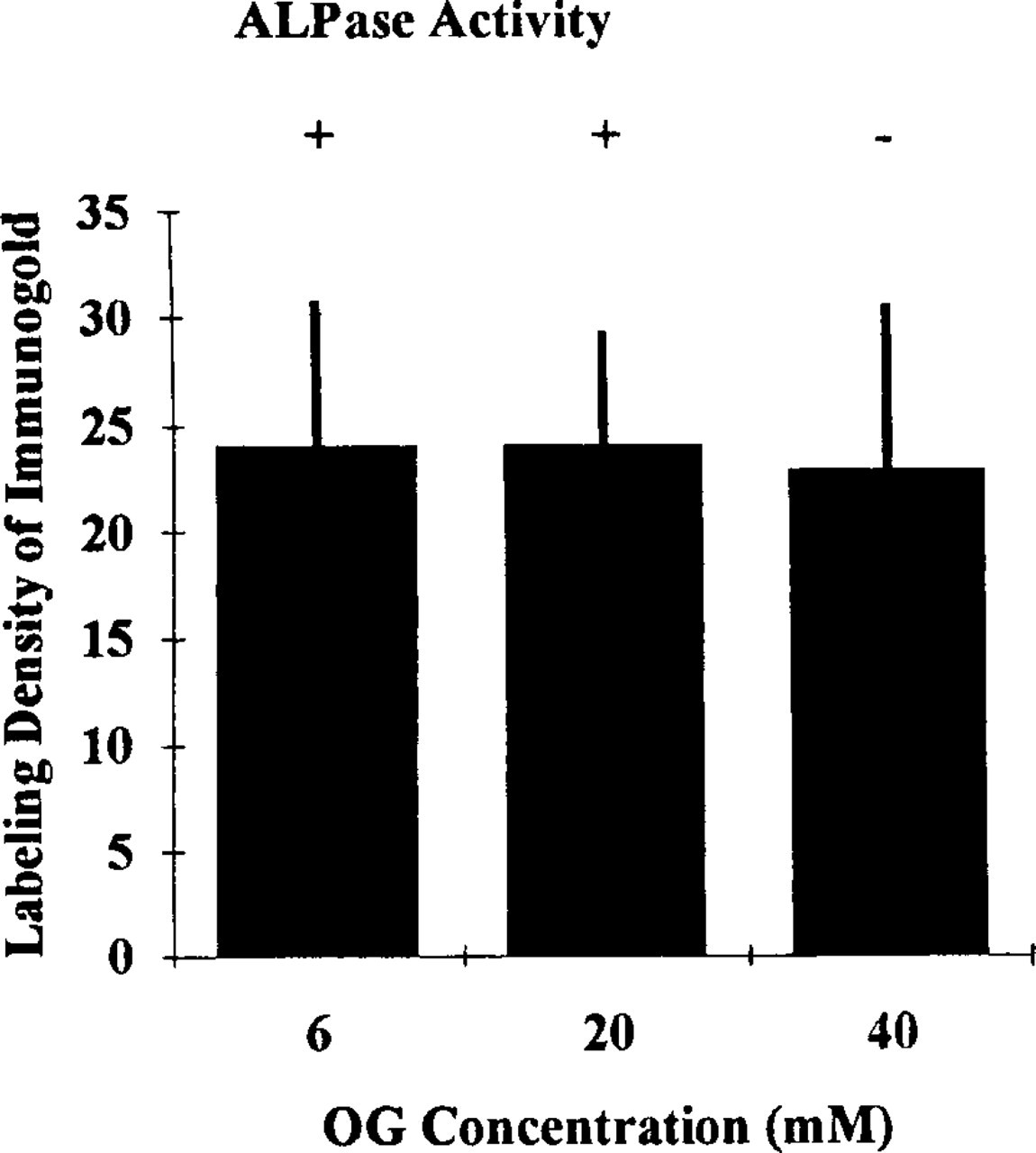
Histogram showing the comparison of labeling density of immunogold probes for HLA Class I by different concentrations of OG. Detection of ALPase activity by OG digestion at each concentration is also indicated: detectable (+) or undetectable (-). Thirty-five micrographs of enlargements of fractured plasma membranes (E-faces) of human neutrophils from each experimental condition were analyzed. The unit of labeling density is the number of gold particles/μm2 of these fractured membranes.
We then combined the immunocytochemical localization of HLA Class I with the enzyme cytochemical localization of ALPase activity on the same replica. The former was found on the plasma membranes of human neutrophils and the latter in the small cytoplasmic granules (Figure 3A). The immunogold particles showing the localization of HLA Class I were restricted to the E-faces of the plasma membranes (Figures 1A and 3A). The cerium reaction product demonstrating ALPase activity was associated predominately with the E-faces of the intracellular granules (Figures 1B and 3A).
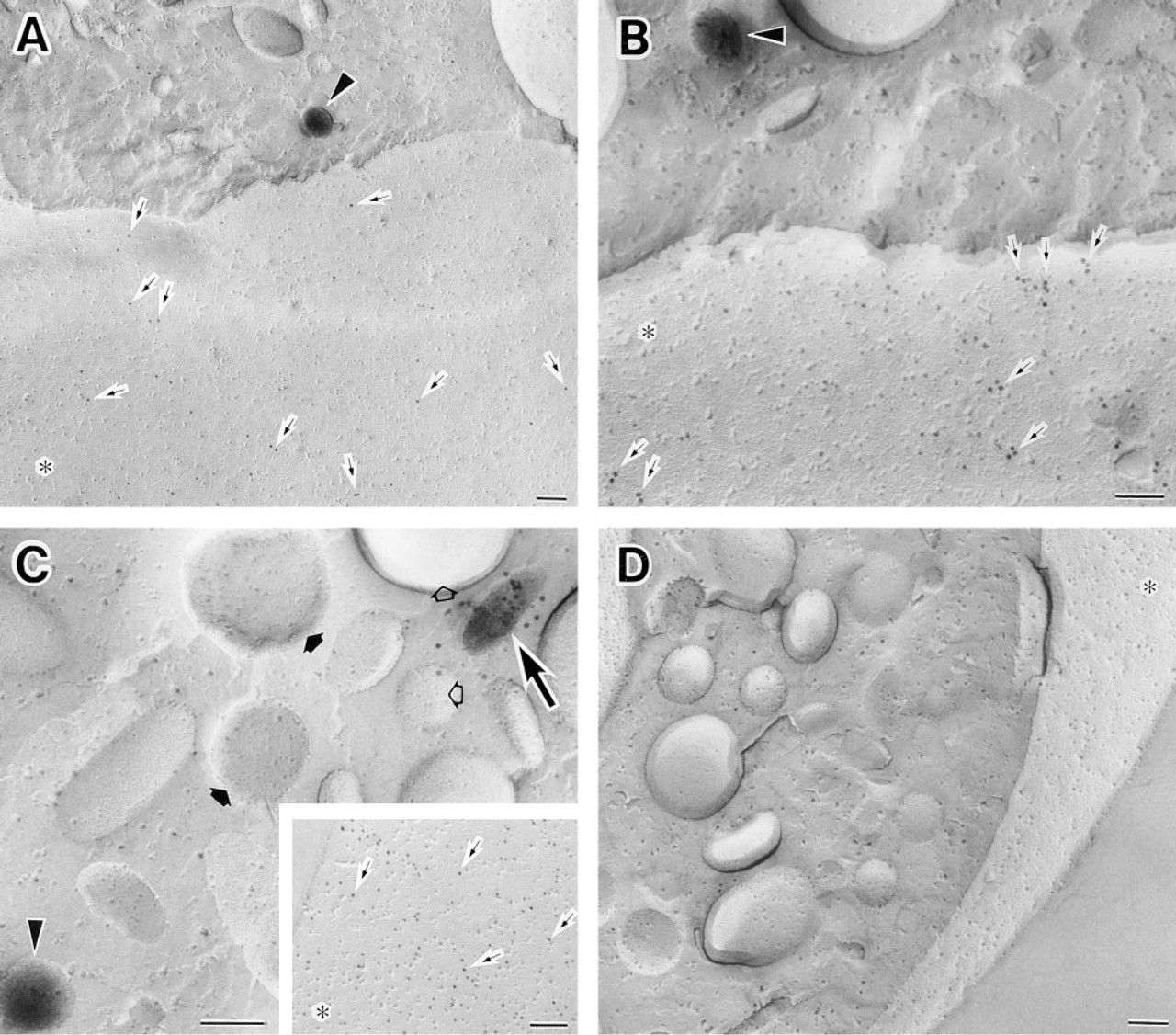
(
Localization of CD62L and ALPase in the same cells was carried out by this new fracture-labeling method. In unstimulated cells, CD62L as well as HLA Class I was demonstrated with 10-nm colloidal gold and was localized on the E-faces of the plasma membranes. ALPase was detected in the cytoplasmic organelles, as mentioned above (Figure 3B). This is essentially consistent with earlier findings suggesting that in unstimulated human neutrophils,
Double labeling of CD16 and ALPase was also achieved on the same replicas (Figure 3C). In unstimulated cells, CD16 was present both on the E-faces of the plasma membranes and the E-faces of cytoplasmic granules. Immunogold particles were sometimes present on the E-faces of the granules that were labeled with the reaction product demonstrating ALPase (Figure 3C). This result suggests heterogeneity in this population of granules (manuscript in preparation). It should be noted that because the electron density of immunogold particles was distinct from that of the cerium phosphate reaction product, co-localization of two different proteins labeled with different cytochemical probes was easily recognized.
Control immunocytochemical incubations consisted of omission of the primary antibodies. Controls for the enzyme cytochemical reaction consisted of adding the ALPase inhibitor levamisole to the complete medium. In each control situation the appropriate labeling was absent (Figure 3D).
Occasionally unfractured cell components were incompletely lysed in the digestion medium and remained attached to the cytochemically labeled replica. These remnants on the replica not only made observation of cell ultrastructure difficult but also induced high background due to the superimposition of the cytochemical probes involved in them on the replica membrane (data not shown). Therefore, the postdigestion after the cytochemical reactions was of importance to overcome this obstacle. We tested many cleaning agents (e.g., OG, SDS, sodium hypochlorite, sodium hydroxide, hydrochloric acid). OG yielded superior results for the postdigestion over any other compound tested.
In this method and in the combined method for cryosections (Takizawa and Robinson 1993), the immunocytochemical procedure was performed first to avoid potential problems of altering or masking antigenic sites owing to the enzyme cytochemical procedure or accumulation of enzyme cytochemical reaction product. However, it was still possible that the second-step cytochemical reaction (i.e., enzyme cytochemical reaction) or the postdigestion could cause dissociation of the antibodies from the antigen sites on replicas. This possibility was tested by a morphometric approach. The labeling density of immunogold particles indicating the distribution of HLA Class I was determined in samples incubated for immunocytochemical localization of HLA Class I alone, after use of the combined approach in which immunocytochemistry was followed by ALPase enzyme cytochemistry, or after that of the postdigestion. Neither the number of colloidal gold particles associated with the plasma membranes after the combined cytochemistry nor that after the post-digestion decreased compared to that after immunocytochemistry alone (Figure 4).
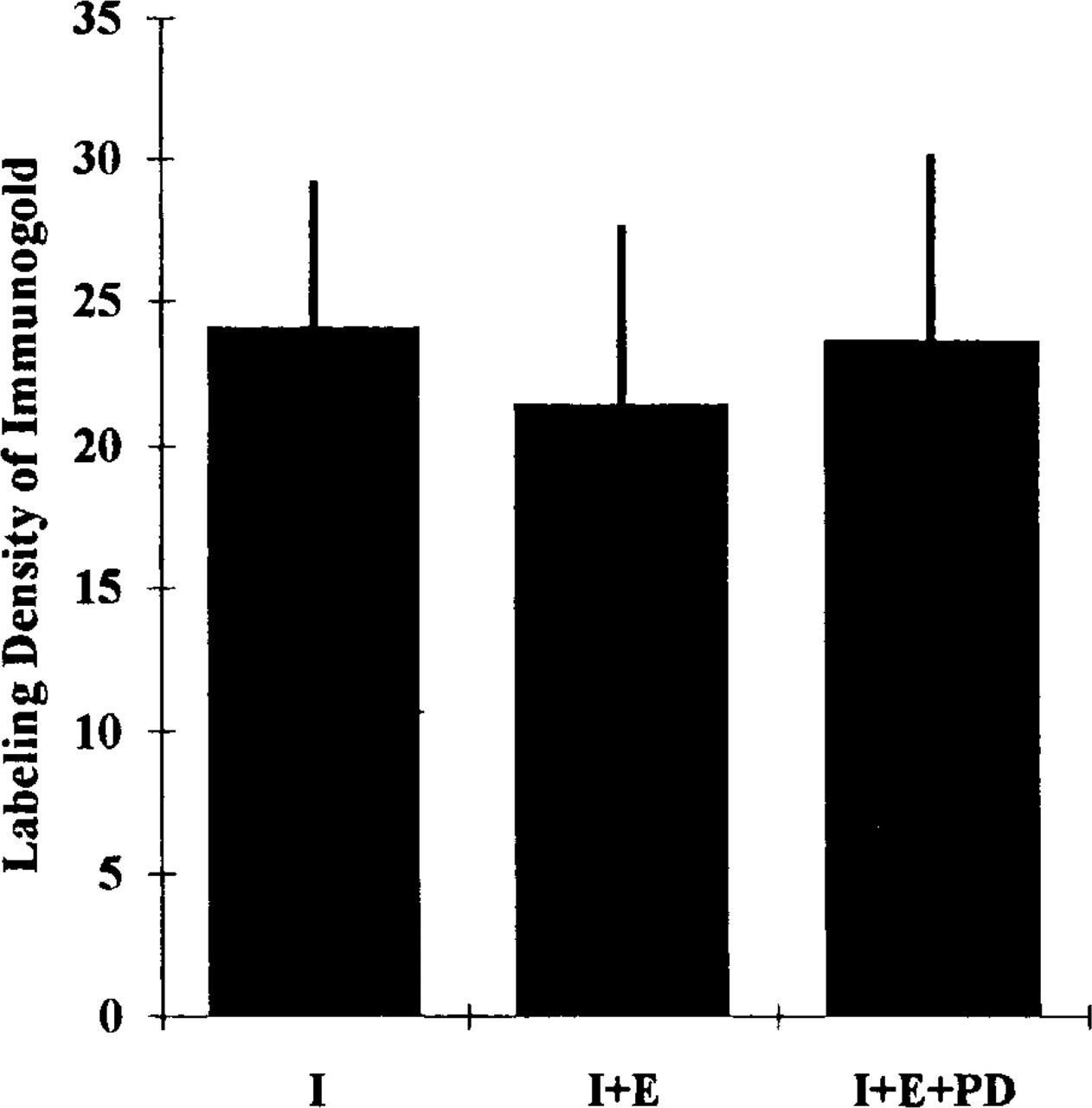
Histogram showing the comparison of labeling density of immunogold probes for HLA Class I between HLA Class I immunocytochemistry alone (I), the combined approach in which HLA class I immunocytochemistry was followed by ALPase enzyme cytochemistry (I + E), and the combined approach followed by postdigestion (I + E + PD). Thirty-five micrographs of enlargements of fractured plasma membranes (E-faces) of human neutrophils from each experimental condition were analyzed. The unit of labeling density is the number of gold particles/μm2 of these fractured membranes.
Discussion
Freeze-fracture immunocytochemistry was first introduced as the “fracture-label” technique for carbohydrate topology in biological membranes (Pinto da Silva et al. 1981a,b,c). Cells were fractured first and then labeled with immunogold probes. This was followed by the development of the “label-fracture” technique, in which cell surfaces were immunocytochemically labeled and subsequently freeze-fractured (Pinto da Silva and Kan 1984). These techniques have been extended further as the “fracture-flip” (Andersson Fersman and Pinto da Silva 1988), the “fracture-flip/Triton X-100” (Fujimoto and Pinto da Silva 1989), and the “simulcast” (Ru-Long and Pinto da Silva 1990) methods to provide high resolution and direct views of the surfaces of biomembranes. However, there have been some technical limitations in these techniques. Thawing before the cytochemical reactions in fracture-label technique has been associated with significant morphological alterations of freeze-fractured membranes and applications of the label-fracture technique have been restricted primarily to cell surfaces. Recently, Fujimoto (1995) reported a new immunocytochemical fracture-label technique to overcome these difficulties. In his experiments, SDS digestion was used to dissolve unfractured cell components before the immunocytochemical reactions that permitted the labeling of antigens in freeze-fractured intracellular membranes. The SDS-digested freeze-fracture replica labeling technique (SDS-FRL) has been applied to studies of epitope topology of intramembranous protein molecules (Fujimoto et al. 1996a) and transmembrane topology of membrane phospholipids (Fujimoto et al. 1996b).
The combination of immunocytochemistry and enzyme cytochemistry is also desirable in certain experimental situations. In our studies, we have used human neutrophils as a model system to investigate the possibility that immunocytochemistry and enzyme cytochemistry could be combined in freeze-fracture cytochemistry. We initially tested the feasibility of SDS-FRL for the double labeling on the same replicas with different cytochemical probes. However, we found that SDS-FRL caused significant enzyme activity loss from Pt/C-stabilized membrane halves (see Figure 1C). It could be speculated that SDS, a strong ionic detergent, completely denatures native enzyme molecule structures, thereby rendering them inactive and unusable for enzyme cytochemical studies. Therefore, in place of SDS, we sought a more suitable compound for replica digestion for this new method. In previous studies, we introduced an enzyme cytochemical fracture-labeling method. ALPase activity was demonstrated on replicas by the use of Triton X-100 or saponin digestion and after ultrasonication treatment (Takizawa and Saito 1997b). However, the immunocytochemical results obtained by these replica cleaning processes were not satisfactory because incompletely digested cell components induced high background and potentially artifactual localization of antigens (data not shown). Previously, we examined the solubilization behavior of GPI-anchored proteins relative to integral membrane proteins in human neutrophils and found that both types of proteins were readily extracted with OG at 4C in unfixed cells (Cain et al. 1995). Therefore, we tested the usefulness of OG digestion for this new method. In the present study, OG digestion was suitable for the combined method and the use of this detergent eliminated several of the problems associated with other detergents. OG is an efficient detergent, and OG digestion preserves both immunocytochemical antigenicity and enzyme activity on the split membrane halves stabilized by Pt/C evaporation. It should be emphasized that stabilization of the fractured membranes with Pt/C evaporation prevents the extraction of membrane proteins in that fracture plane. Our data indicate that OG digestion is crucial to the success of the combined cytochemistry on the replicas of human neutrophils.
OG, a nonionic detergent, was initially reported as an effective agent for solubilizing membrane proteins (Baron and Thompson 1975). In biochemical studies, the advantages of the use of OG are not only the stability of biological activity of membrane proteins in OG but also the ability to readily remove this detergent by gel filtration chromatography and dialysis (Baron and Thompson 1975). This ability is due to the high critical micelle concentration (CMC) of OG. The CMC of OG is 20–25 mM, whereas that of Triton X-100 is only 0.24 mM (Shinoda et al. 1961; Helenius and Simons 1975; Gould et al. 1981). It appears that the CMC of detergents affects enzyme cytochemical detection as well as the biochemical solubilization of membrane proteins. However, concentrations of OG greater than 20 mM made detection of enzyme activity on replicas difficult (see Figure 2). Nevertheless, it did not affect the immunocytochemical detection of proteins. Concentrations as high as 40 mM OG preserved antigenicity on replicas (see Figure 2). We recommend that OG for the replica digestion before cytochemical incubations be used at the CMC (i.e., about 20 mM).
The order of the reactions is important. The immunocytochemical localization is always the first step, to eliminate the probability of interference from the enzyme cytochemical reactions. The enzyme cytochemical reaction may modify the efficiency of the subsequent immunolocalization. This might be a particular problem when co-localization of the enzyme cytochemical and immunocytochemical products occurs. The metal precipitations from enzyme cytochemical reaction may cover antigenic sites or in some other ways inhibit antigen-antibody binding. In the present study, immunocytochemistry followed by enzyme cytochemistry showed the co-localization of CD16 and ALPase on the same intracellular granule (see Figure 3C). Therefore, we have developed a procedure in which combined immunocytochemistry and enzyme cytochemistry can be conducted without these potential complications.
In our experiments, it was still possible that the subsequent cytochemical reaction and the postdigestion might lead to loss of immunolabeling from the replicas. This was tested by morphometric analysis of the immunogold labeling and was found not to occur (see Figure 4). In other applications of this combined approach, similar control experiments would be necessary to determine whether or not the particular enzyme cytochemical reaction employed affects immunocytochemical labeling. In addition, the enzyme cytochemical reaction product (i.e., cerium phosphate) is not affected by OG digestion. Indeed, it is stable after treatment with household bleach, one of the most powerful replica cleaning agents (Takizawa and Saito 1996). To the best of our knowledge, a technique combining immunocytochemistry and enzyme cytochemistry on the same replicas has not been reported.
Combined immunocytochemical and enzyme cytochemical reactions on the same replicas would have many applications and should provide new and important information that may be difficult to obtain by other approaches, e.g., the localization of adhesion molecules and glycosyl-phosphatidylinositol (GPI)-anchored proteins in human neutrophils. It has been recently suggested that adhesion molecules (e.g., CD11b, CD62L) and ALPase, which is a GPI-anchored protein, simultaneously change these subcellular localizations and surface expressions in human neutrophils during stimulation (Borregaard et al. 1994). However, the high-resolution morphological relationship between these molecules remains to be determined. In this study, we show the co-localization of two different GPI-anchored proteins in the same granule (see Figure 3C). We are presently studying the dynamics of intracellular granules containing GPI-anchored proteins and adhesion molecules in neutrophils in greater detail by this new methodology (unpublished observations). In this method, the fixation for cytochemical detection of membrane proteins is stabilization by Pt/C evaporation (i.e., physical fixation), so that this method may be particularly useful for detection of the proteins whose biological activities (i.e., antigenicity and enzyme activity) are susceptible to inhibition by chemical fixation (e.g., glutaraldehyde and/or paraformaldehyde fixation).
In conclusion, we show a new freeze-fracture cytochemistry combining immunocytochemistry and enzyme cytochemistry. This technique enables us to reveal the relationship between molecules in biological membranes by double labeling with two different cytochemical markers (i.e. immunogold particles and cerium phosphate reaction product). This technique should be a useful addition for studying the ultrastructural organization of biomembranes.
Footnotes
Acknowledgements
Supported by grants-in-aid for Scientific Research (nos. 08670030 and 09770016) from the Ministry of Education, Science, Sports and Culture of Japan (TS, TT), by a grant from the Kazato Research Foundation (TT), and by a grant from the Nippon Foundation (TT).
We thank Dr Clark L. Anderson for his generous gift of antibody. We are deeply indebted to Ms Kiyomi Inose, Ms Kaori Ishikawa, Ms Chiaki Ishijima, and Ms Megumi Yatabe for excellent technical assistance.