
Abstract
Various CC chemokine receptors are expressed on effector cells in allergic inflammation and their distinct expression pattern may dictate, to a large extent, the migration of inflammatory cells to sites of airway inflammation. The lipopolysaccharide (LPS)-inducible CC chemokine receptor (L-CCR) is an orphan chemokine receptor that has previously been identified in the murine macrophage cell line RAW 264.7 and in murine brain glial cells. In this study we investigated the induction and localization of L-CCR mRNA expression in mouse lung after ovalbumin (OVA)-induced airway inflammation. Both RT-PCR experiments and in situ hybridization (ISH) experiments in whole lung sections revealed a rapid upregulation of L-CCR mRNA expression as early as 1 hr and 3 hr after OVA challenge. Expression was found predominantly in MAC3+ macrophages and in bronchial epithelium, as shown by ISH and immunohistochemistry (IHC). We demonstrated that L-CCR mRNA expression is strongly upregulated in mouse lung after OVA challenge and is localized in macrophages and bronchial epithelium. Regarding the likely role of L-CCR as a chemokine receptor with the putative ligand monocyte chemotactic protein-1 (MCP-1, CCL2), this receptor may have an important function in the early phase of airway inflammation.
C
The immune response associated with allergic airway inflammation is characterized by infiltration of the airways with eosinophils, basophils, lymphocytes, neutrophils, and macrophages (Lukacs et al. 1995). Several CC chemokine receptors, i.e., CCR2, CCR3, CCR4, CCR6, and CCR8, have been associated with effector cells in allergic inflammation, and their distinct expression is crucial for the migration of inflammatory cells to sites of allergic airway inflammation (D'Ambrosio et al. 2001; Lukacs 2001). Knockout mouse models have provided further evidence of the importance of CC chemokine receptor signaling in allergic airway inflammation. Disruption of one of the above-mentioned CC chemokine receptors was associated with attenuated eosinophil recruitment to the lung (CCR3, CCR4, CCR6, and CCR8) (Chensue et al. 2001; Lukacs et al. 2001; Humbles et al. 2002; Schuh et al. 2002), reduced Th2-type cytokine levels (CCR4, CCR6, and CCR8) (Chensue et al. 2001; Lukacs et al. 2001; Schuh et al. 2002), and reduced airway hyperresponsiveness (CCR4 and CCR6) (Lukacs et al. 2001; Schuh et al. 2002). Surprisingly, CCR3–/– knockout mice showed increased airway hyperresponsiveness with no changes in Th2 cytokine levels (Humbles et al. 2002). With respect to the development of pulmonary eosinophilia and airway hyperresponsiveness during allergen-induced airway inflammation, conflicting results have been obtained with CCR2–/– knockout mice (Campbell et al. 1999; Blease et al. 2000; MacLean et al. 2000; Kim et al. 2001). Interestingly, the CCR2 ligand monocyte chemotactic protein-1 (MCP-1, CCL2) is notably increased in murine airways after allergen challenge (Gonzalo et al. 1996), and neutralization of CCL2 resulted in clearly reduced accumulation of eosinophils, coinciding with reduced airway hyperresponsiveness and reduced IgE and IL-4 levels (Gonzalo et al. 1998). In humans, ten CC chemokine (CCRs) and five CXC chemokine receptors (CXCRs) have been cloned thus far (Murphy et al. 2000). In addition, a variety of orphan receptors with chemokine-like structures have been described (see databases such as www.GPCR.org).
The orphan chemokine receptor lipopolysaccharide (LPS)-inducible CC chemokine receptor (L-CCR) was first characterized in the mouse macrophage cell line RAW 264.7 (Shimada et al. 1998). Recently, RT-PCR and in situ hybridization (ISH) experiments revealed L-CCR mRNA expression in both mouse microglia and astrocytes (Zuurman et al. 2003). Similar to results obtained with the RAW 264.7 cell line (Shimada et al. 1998), L-CCR mRNA expression was enhanced under proinflammatory conditions (Zuurman et al. 2003). Because L-CCR mRNA expression has also been found in mouse lung tissue (Biber et al. 2003), we investigated the induction and localization of L-CCR mRNA expression in the lung in a murine model of ovalbumin (OVA)-induced airway inflammation.
Materials and Methods
Antibodies and Reagents
MAC3 and GR1 rat anti-mouse MAbs were purchased from Pharmingen (San Diego, CA). Goat polyclonal antibody against CCR2 (C20) was purchased from Santa Cruz Technology (Santa Cruz, CA). Taq-polymerase was purchased from InViTek (Berlin, Germany). TA vectors pCR2.1 and pCRII were obtained from Invitrogen (Breda, The Netherlands). Digoxigenin-conjugated UTP and alkaline phosphatase (AP)-conjugated sheep anti-digoxigenin were from Boehringer Mannheim (Mannheim, Germany). Ovalbumin, 50 × Denhardt's solution, and RNase were purchased from Sigma (St Louis, MO). Biotinylated goat anti-rat F(ab)2 fragments were from Southern Biotechnology Associates (Birmingham, AL) and biotinylated rabbit anti-goat immunoglobulins were purchased from DAKO (Glostrup, Denmark).
OVA Challenge
Two weeks before the in vivo procedures, 8–10-week-old male C57BI/6-J mice were obtained from Harlan (Heath-field, UK). Mice were challenged by a modification of the challenge procedure described by Gonzalo et al. (1996). In brief, mice were sensitized intraperitoneally (IP) with OVA (0.1 mg/mouse) in PBS at day 0, followed by 5 min of inhalation with a 2% OVA aerosol in PBS at day 8, using specially designed perspex cages with an internal volume of 9 liter, in which the mice could move freely. For control experiments, a second group of mice inhaled PBS instead of OVA. For allergen provocation, mice were repetitively challenged for 20 min with a 1% OVA aerosol in PBS once a day at days 15–18. Control mice were challenged for 20 min with PBS. Mice were sacrificed 1, 3, or 6 hr after the last OVA challenge and 3 hr after the last PBS challenge. Both lungs of the challenged mice were dissected and immediately placed in liquid nitrogen. The study was approved by the University of Groningen Committee for Animal Experimentation, which is responsible for the care and proper use of experimental animals.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Mouse lung tissue was lysed in guanidinium isothiocyanate/mercaptopropanol buffer and total RNA was extracted with slight modifications according to Chomczynski and Sacchi (1987). Total RNA was transcribed into cDNA and amplified by PCR as described (Biber et al. 1997), using primer pairs for CCL2, L-CCR, and GAPDH, respectively. PCR products were size-fractionated on a 1.5% agarose gel and mRNA expression was determined relative to GAPDH mRNA expression. Potential contamination by genomic DNA was checked by running the reverse transcription reactions without reverse transcriptase and using GAPDH primers in subsequent PCR amplifications. Primer sequences, cycle numbers, and annealing temperatures are listed in Table 1. Cloning into a PCR2.1 vector and subsequent sequencing checked the identity of all PCR products.
In Situ Hybridization (ISH)
ISH was performed as described (Copray and Brouwer 1994). In brief, the L-CCR PCR product was cloned into the dual promoter PCR II vector and linearized. L-CCR sense and antisense probes were synthesized by run-off transcription and the use of digoxigenin-conjugated UTP according to the manufacturer's protocol (Boehringer Mannheim). Lung tissue sections (4 μm) were incubated in 4% paraformaldehyde, rinsed with potassium phosphate buffered saline (KPBS), and digested for 30 min in maleate buffer containing 0.5% Triton. Then lung sections were rinsed in 2 × SSC (1 × SSC = 150 mM NaCl, 15 mM Na-citrate), dehydrated in an ethanol series, and dried by air. Sections were hybridized overnight at 60C in a solution containing 50% formamide, 0.3 M NaCl, 10 mM Tris (pH 8.0), 1 mM EDTA, 0.05% tRNA, 1 × Denhardt's solution, and 10% dextran sulfate. Final probe concentrations in hybridization buffer were 1.5 ng/μl. After hybridization the sections were treated with 10 μg/ml of ribonuclease A for 30 min at 37C and washed in 0.1 × SSC at 65C. The immunological detection of the digoxigenin-labeled RNA-RNA complex was preceded by 30 min of preincubation at room temperature (RT) in 0.1 M Tris, 0.15 M NaCl, pH 7.5 (buffer 1) containing 5% BSA. Sections were incubated for 2 hr at RT with the AP-conjugated sheep anti-digoxigenin diluted 1:500 in buffer 1 containing 1% BSA. After thorough rinsing in buffer 1 and a 10-min preincubation in an alkaline buffer solution (ABS: 0.1 M Tris, 0.1 M NaCl, 0.05 M MgCl2.6H2O, pH 9.5), the AP activity was revealed with a freshly prepared solution of 0.34 mg/ml nitroblue tetrazolium and 0.17 mg/ml 5-bromo-4-chloro-3-indolyl phosphate in ABS. Endogenous phosphatase activity was inhibited by the addition of levamisole (0.24 mg/ml) to the staining solution. The color development was terminated after approximately 75 min by placing the sections in a buffer solution consisting of 0.01 M Tris, 1 mM EDTA, pH 8.5. For analyzing constitutive L-CCR mRNA expression, color development was terminated after 3–4 hr. The dark-purple precipitate, indicating the presence of hybridized mRNA, was visualized with brightfield microscopy. Control experiments included hybridization with digoxigenin-labeled sense probes and hybridization after treatment of the sections with RNase for 30 min.
Primers used for mouse CCL2 (formerly known as MCP-1), L-CCR and GAPDH

Immunohistochemistry
Frozen lung sections were dried and incubated in acetone for 10 min. For immunodetection of macrophages, neutrophils, and CCR2, lung sections (4 μm) were incubated with MAC3, GR1 MAbs and a goat polyclonal antibody against CCR2 (C20), respectively. Staining was detected using biotinylated goat anti-rat F(ab)2 fragments (MAC3 and GR1) and biotinylated rabbit anti-goat immunoglobulins (CCR2), followed by incubation with AP-labeled streptavidin. The AP activity was revealed by 1 mg/ml Fast Red staining in 0.1 M Tris-HCl (pH 8.2) containing 0.2 mg/ml naphthol AS-MX, 0.3% MgSO4, and 0.24 mg/ml levamisole to inhibit endogenous phosphatase activity. Eosinophils in lung tissue were revealed by cyanide-resistant diaminobenzidine (DAB) staining. Measurement of numbers of eosinophils in lung tissue was done by computer-assisted morphometric analysis and expressed as volume percent of tissue.
Results
Regulation of L-CCR mRNA Expression in Mouse Lung After OVA Challenge
OVA challenge was performed as described in Materials and Methods. For RT-PCR analysis and ISH experiments, both lungs were removed at 1, 3, or 6 hr after the last of four consecutive daily challenges with OVA and at 3 hr after challenges with PBS as a control. Compared to the lungs of PBS-challenged mice (Figure 1A), eosinophils were largly increased at 6 hr after OVA challenge (Figure 1B). Whereas neutrophils were not detectable in lung sections of PBS-challenged mice (Figure 1C), they were numerous at 1 hr after OVA challenge (Figure 1D). Using a similar challenge protocol, Gonzalo et al. (1996) described an increase in CCL2 mRNA expression. We therefore used OVA-induced CCL2 mRNA expression, as observed with RT-PCR, as an additional positive control in our challenge protocol (Figure 1E). Compared to the lungs of PBS-challenged mice, L-CCR mRNA expression had increased after OVA challenge. Semiquantitative analysis revealed a strong increase in L-CCR mRNA expression 1 and 3 hr after OVA, whereas L-CCR mRNA expression had returned to control levels at 6 hr after OVA challenge. GAPDH mRNA expression was similar at all time points (Figure 1E).
Results obtained with RT-PCR were verified by ISH. Compared to lung sections of PBS-challenged mice (Figure 2A), an upregulation of L-CCR mRNA expression was visible 1 and 3 hr after OVA challenge (Figures 2B and 2C). Sense staining in a sequential lung section 3 hr after OVA challenge was clearly negative (Figure 2D), indicating specificity of the antisense signal. Similar to the results obtained by RT-PCR, L-CCR mRNA expression declined 6 hr after OVA challenge in two of three separate experiments (data not shown). Whereas ISH conditions in the above-mentioned experiments were chosen to detect an induction of L-CCR mRNA expression, more prolonged staining revealed constitutive expression of L-CCR mRNA in lung tissue 3 hr after PBS challenge, as shown in Figure 3E. Prolonged staining of lung tissue 3 hr after PBS challenge with the sense L-CCR probe was clearly negative (Figure 3F).
Localization of L-CCR mRNA Expression in Mouse Lung After OVA Challenge
Results obtained with both RT-PCR and ISH analysis indicated an increase in L-CCR mRNA expression in mouse lung after OVA challenge. Additional experiments were performed to identify which cell types express L-CCR mRNA. It is known that the cellular infiltrate in mouse lung after OVA challenge consists of eosinophils, lymphocytes, macrophages, and neutrophils (Blyth et al. 1996). Because L-CCR mRNA expression has recently been reported in macrophage-like cells (Shimada et al. 1998; Zuurman et al. 2003), we performed L-CCR ISH combined with specific immunohistochemistry (IHC) of macrophages using an MAC3 MAb. Because, for technical reasons, double staining was not possible, we analyzed L-CCR mRNA expression and MAC3 staining in serial lung sections. MAC3 staining (Figure 3A), together with ISH of L-CCR mRNA in directly adjacent lung sections (Figure 3B), clearly shows L-CCR mRNA expression in macrophages 3 hr after OVA challenge. L-CCR mRNA also localized in macrophages 1 hr after OVA challenge (data not shown).
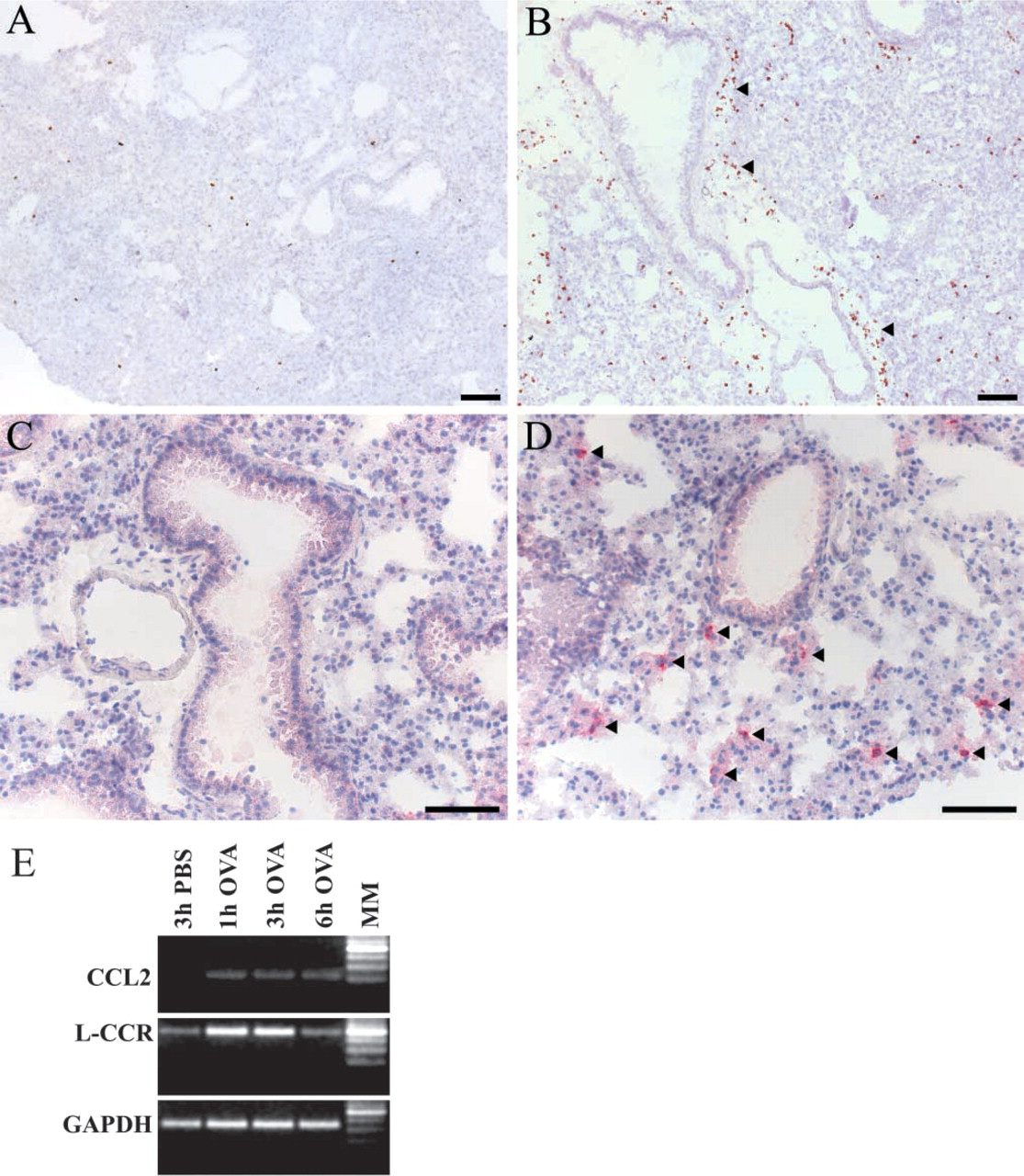
IHC staining (
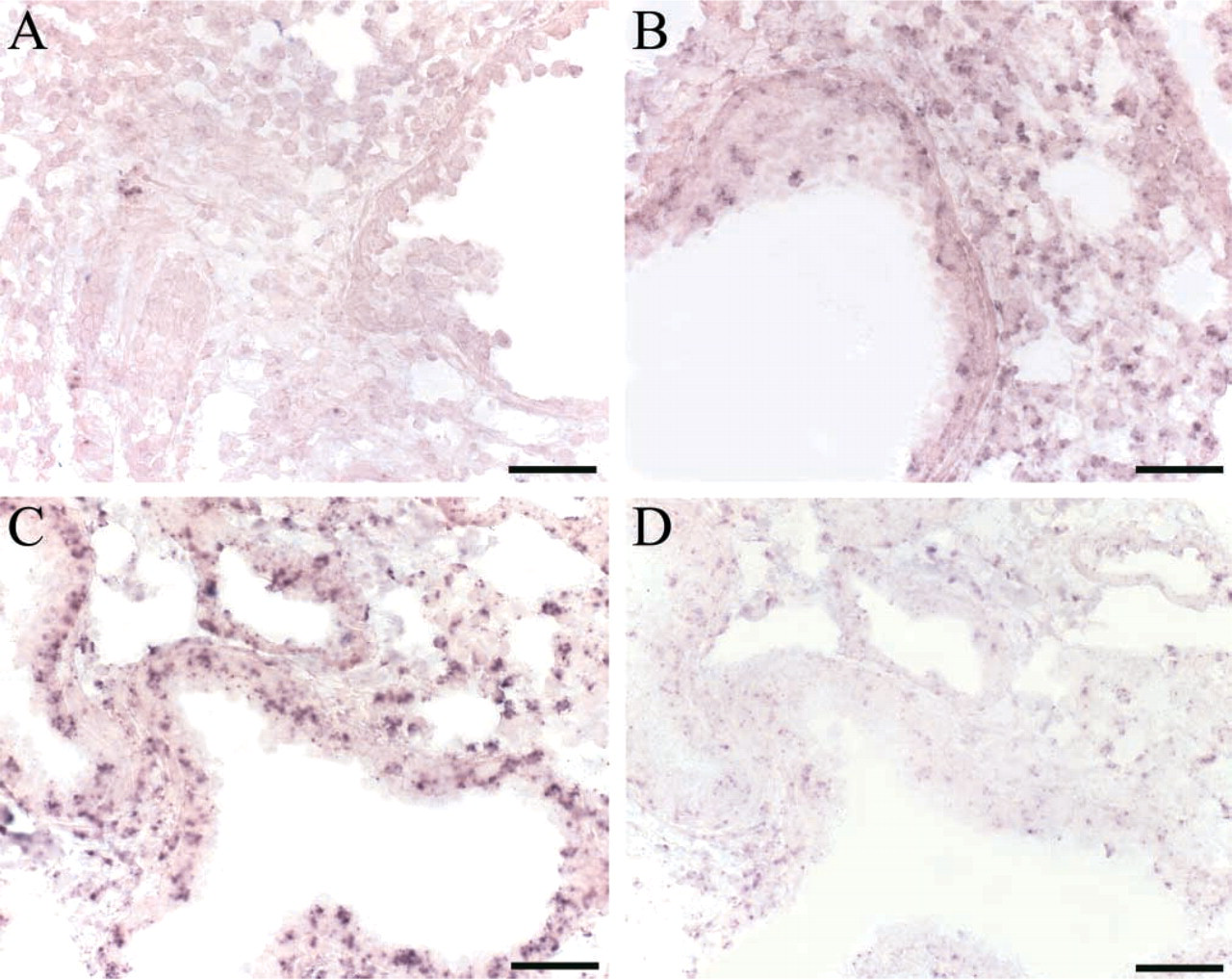
Representative L-CCR mRNA ISH in frozen lung sections from mice obtained 1 or 3 hr after the last of four daily challenges with 1% OVA and 3 hr after the last of four daily challenges with PBS. (
Because it was apparent in Figures 3A and 3B that L-CCR mRNA expression was not found only in macrophages, we evaluated localization of L-CCR mRNA in other cell types. ISH experiments showed strong expression of L-CCR mRNA in bronchial epithelium at both 3 hr (Figure 3B) and 1 hr (Figure 3D) after OVA challenge. Moreover, prolonged staining revealed constitutive L-CCR mRNA expression in bronchial epithelium, as observed in lung tissue 3 hr after PBS challenge (Figure 3E). We also performed IHC with MAb GR1 to stain neutrophils (Figure 3C) and L-CCR ISH in sequential lung sections (Figure 3D). Again, double staining was not possible for technical reasons. The timepoint and localization of L-CCR mRNA expression suggest L-CCR mRNA expression in neutrophils 1 hr (Figures 3C and 3D) and 3 hr (data not shown) after OVA challenge. In addition, the presence of infiltrated eosinophils was examined. Because eosinophils in our model were increased only at 6 hr after OVA challenge (Figures 1B and 4) and L-CCR mRNA expression was normalized at this timepoint (Figure 1E), no expression of L-CCR mRNA was present in eosinophils.
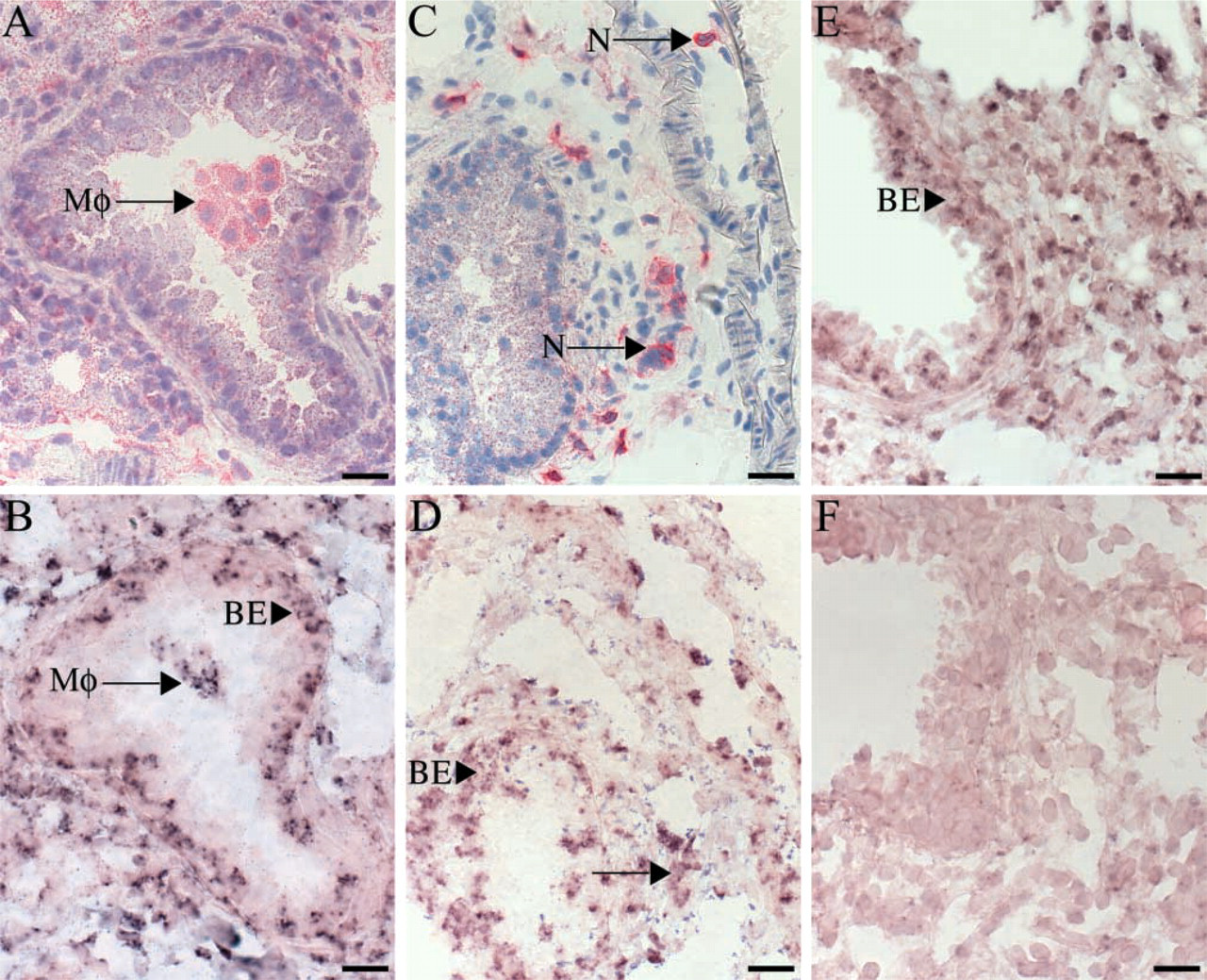
L-CCR mRNA ISH and IHC in sequential lung sections. (
Localization of CCR2 Protein in Mouse Lung
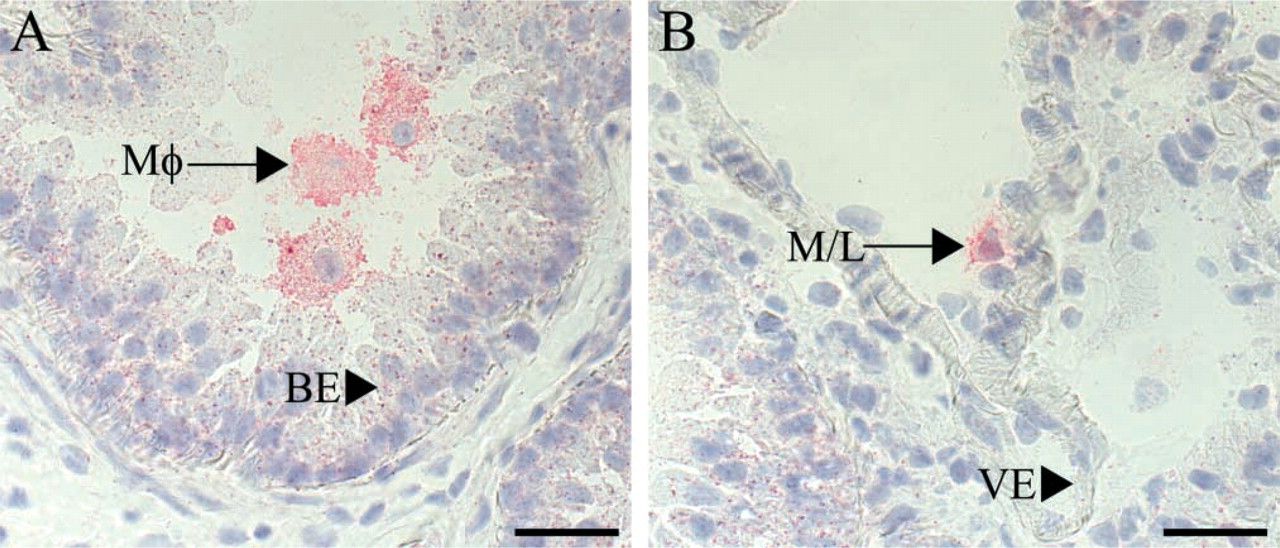
L-CCR mRNA was expressed in murine bronchial epithelium and in lung macrophages. Moreover, we confirmed the previously observed OVA-induced CCL2 mRNA in mouse lung tissue (Gonzalo et al. 1996). The only identified CCL2 receptor is CCR2, which is also highly expressed on monocytes/macrophages (Murphy et al. 2000). Accordingly, mouse models with target disruption of CCR2 have identified CCR2 to be important for monocyte/macrophage recruitment to sites of inflammation (Boring et al. 1997; Kurihara et al. 1997). To determine CCR2 protein expression in mouse lung, IHC with a goat polyclonal antibody against CCR2 (C20) crossreacting with mouse CCR2 was performed. Clear CCR2 expression was seen on macrophages in both PBS- (control) and OVA-challenged animals at all time points. Representative positive staining of macrophages at 1 hr after OVA challenge is shown in Figure 5A. In addition, CCR2-positive cells were found attached to the vascular endothelium 6 hr after OVA challenge, probably representing monocytes or lymphocytes (Figure 5B). No expression of CCR2 was identified on either bronchial epithelium or vascular endothelium in mouse lung at any time point, as shown in Figures 5A and 5B.
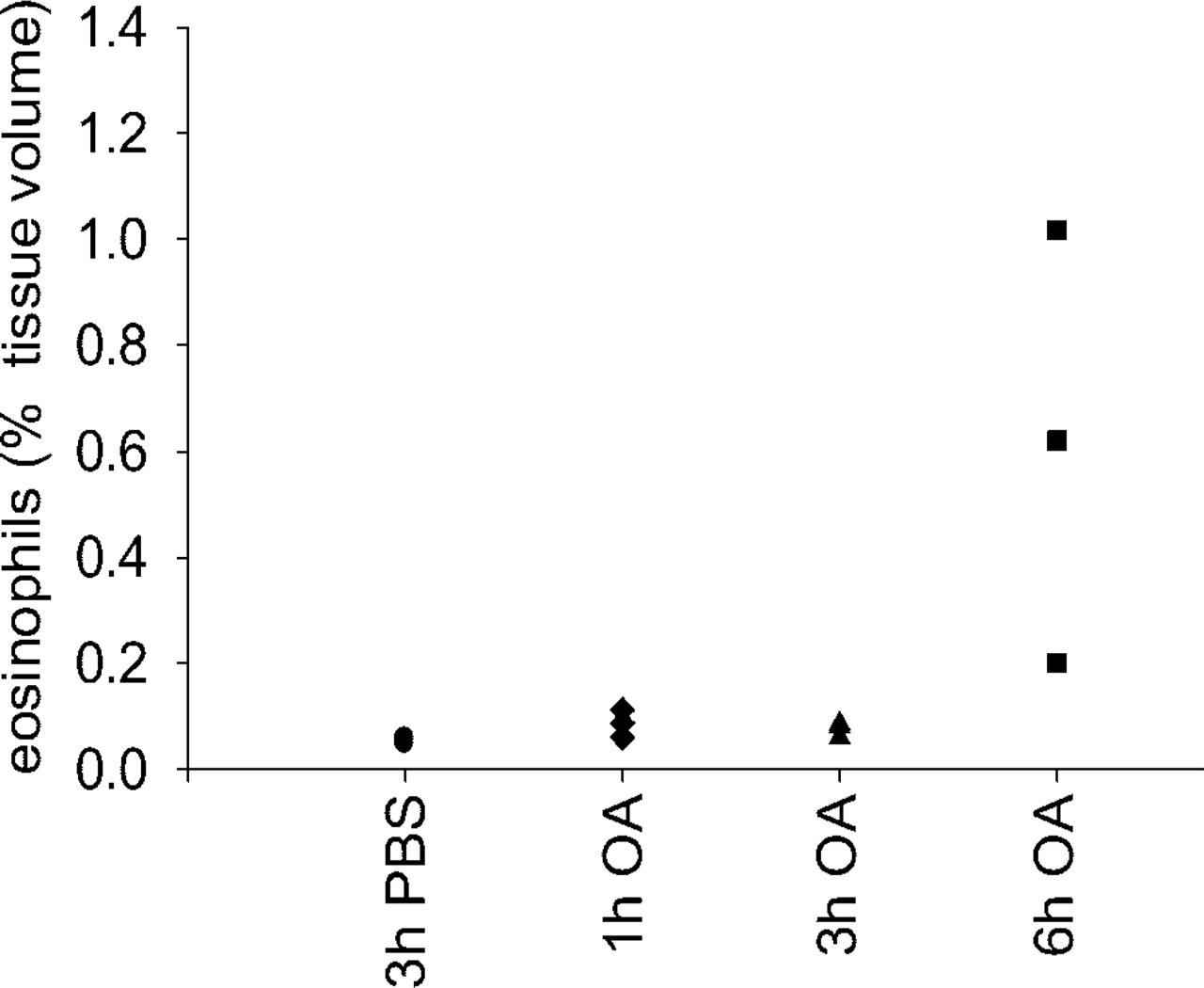
Eosinophils, expressed as volume percent in whole tissue as revealed by computer-assisted morphometric analysis. Eosinophils were revealed by cyanide-resistant DAB staining of frozen lung sections dissected from mouse 3 hr after PBS (control; circles, n = 3) and 1 hr (diamonds, n = 3), 3 hr (triangles, n = 3) and 6 hr (squares, n = 3) after OVA challenge.
Discussion
Because L-CCR mRNA has been detected in murine lung (Biber et al. 2003) and its expression was induced in a proinflammatory response (Shimada et al. 1998; Zuurman et al. 2003), we have investigated its induction and localization of expression in the lung in a murine model of OVA-induced allergic airway inflammation. The results showed a rapid upregulation of L-CCR mRNA expression in mouse lung after OVA challenge. This L-CCR mRNA expression was clearly localized in lung macrophages and in bronchial epithelium.
Our results revealed a rapid upregulation of L-CCR mRNA in whole lung tissue after OVA challenge. At present, little is known about allergen-induced expression of CC chemokine receptors. An allergen-induced increase of CCR4 protein (Panina-Bordignon et al. 2001) and mRNA (Nouri-Aria et al. 2002) expression has been observed in lung tissue of atopic asthmatics, whereas CCR4 mRNA was undetectable in the lungs of non-asthmatic subjects both before and after allergen challenge (Nouri-Aria et al. 2002). In addition, allergen provocation studies performed on mice have revealed allergen-induced expression of CCR2 and CCR5 on macrophages (Wells and Proudfoot 1999). In accordance with the latter observation, we have found a rapid induction of CCR2 mRNA in mouse lung after OVA challenge by RT-PCR experiments in whole lung sections (data not shown).
In our study, we found L-CCR mRNA expression in MAC3+ mouse lung macrophages. This expression is in accordance with results obtained by Shimada et al. (1998) in the murine macrophage RAW264.7 cell line. CCR1, CCR2, and CCR5 proteins are expressed on human monocytes/macrophages (Murphy et al. 2000). Similar expression levels of CCR2 and CCR5 proteins have been identified on murine monocytes/macrophages (Mack et al. 2001), and CCR2 is expressed on mouse resident alveolar macrophages (Maus et al. 2002). In accordance, we also found CCR2 protein to be expressed on mouse lung macrophages. Furthermore, the orphan CC chemokine receptor human chemokine receptor (HCR) has recently been identified on human monocytes/macrophages (Migeotte et al. 2002). The role played by alveolar macrophages in allergic lung inflammation pertains to their ability to serve as an antigen-presenting cell. More important, however, is their ability to produce pro- and antiinflammatory cytokines, lipid mediators, radical oxygen species, and chemokines (Lukacs et al. 1995; Jansen 1996; Gosset et al. 1999). Chemokine receptors expressed on monocytes/macrophages not only play a crucial role in the selective recruitment of these cells to sites of inflammation (Kurihara et al. 1997), but are also important in the regulation of adhesion molecule expression and the release of inflammatory mediators by monocytes/macrophages (Jiang et al. 1992; Mackay 2001). In addition to L-CCR mRNA expression in macrophages, our results suggest L-CCR expression in neutrophils. Unlike the more easily identified macrophages, mostly lying in clusters without many admixed cells, neutrophils were scattered. Therefore, it was not possible to be sure about the exact localization of L-CCR mRNA expression in neutrophils in sequential lung sections. Further experiments using a purified neutrophil population from mouse lung would be needed to confirm L-CCR mRNA expression in neutrophils. Whereas L-CCR mRNA was rapidly induced and observed at 1 and 3 hr after OVA challenge, eosinophils were selectively increased at 6 hr after OVA challenge and showed no L-CCR mRNA expression.
In human asthmatics, CCL2 (formerly known as MCP-1) expression in the airways is notably increased (Sousa et al. 1994; Alam et al. 1996). The rapid upregulation of CCL2 mRNA observed in our murine model of OVA-induced airway inflammation confirms findings by Gonzalo et al. (1996), who have also shown that CCL2 mRNA expression correlates with the accumulation of monocytes/macrophages in the lung (Gonzalo et al. 1998). Moreover, neutralization of CCL2 resulted in reduced accumulation of monocytes/macrophages, T-lymphocytes, and eosinophils, coinciding with reduced IgE and IL-4 levels in the bronchoalveolar lavage and reduced airway hyperresponsiveness (Gonzalo et al. 1998). In addition, CCL2-deficient mice have impaired monocyte trafficking (Lu et al. 1998) and are unable to mount Th2 cytokines (IL-4 and IL-5), suggesting a role for CCL2 in Th2 polarization (Gu et al. 2000). CCR2 is the only CCL2 receptor identified thus far (Murphy et al. 2000). Although both CCL2- and CCR2-deficient mice show defects in monocyte/macrophage recruitment (Boring et al. 1997; Kurihara et al. 1997; Lu et al. 1998), conflicting results have been obtained in CCR2–/– knockout mice with respect to the development of pulmonary eosinophilia and airway hyperresponsiveness during allergen-induced airway inflammation (Campbell et al. 1999; Blease et al. 2000; MacLean et al. 2000; Kim et al. 2001). Therefore, differential use of CCR2 by multiple ligands or the existence of a second CCL2 receptor has been hypothesized (Lukacs 2001). Cultured murine microglia highly express L-CCR mRNA and show biotinylated CCL2-binding signals, CCL2-induced intracellular calcium mobilization, and CCL2-promoted chemotactic response in the absence of detectable CCR2 mRNA expression (Zuurman et al. 2003). In addition, expression of L-CCR in HEK 293 cells revealed functional chemokine responses to CCL2 (Biber et al. 2003). It is therefore tempting to speculate that L-CCR is a functional chemokine receptor (Zuurman et al. 2003). Because brain microglia show many macrophage-like properties, we speculate that L-CCR could contribute to the activation of lung macrophages and the recruitment of monocytes to murine lung, possibly via CCL2 binding. Obviously, target disruption of L-CCR in a murine model of airway inflammation will be necessary to reveal a possible role of L-CCR in leukocyte recruitment or activation during airway inflammation.
IHC staining of frozen lung sections with a goat polyclonal antibody against CCR2 (C20). (
Interestingly, ISH experiments revealed L-CCR mRNA expression in the bronchial epithelium. Recent reports describe functional CC chemokine receptors on structural cells, such as smooth muscle cells (Schecter et al. 2000) and endothelial cells (Salcedo et al. 2000). Positive CCR3 staining of the airway epithelium has been observed in biopsies from asthmatic subjects, and stimulation with the CCR3 agonist CCL11 (formerly known as eotaxin) revealed a functional receptor on the human bronchial epithelial BEAS-2B cell line (Stellato et al. 2001). Moreover, the CXC chemokine receptor CXCR4 has also shown to be functionally expressed on the alveolair epithelial A549 cell line (Murdoch et al. 1999) and on primary human bronchial epithelial cells (Eddleston et al. 2002). Because the epithelial cell is a main source of chemokines, an autoregulatory function for chemokine receptors on the epithelial cell has been hypothesized (Stellato et al. 2001). L-CCR might be a functional chemokine receptor on the mouse bronchial epithelium. With regard to the latter hypothesis, it is interesting that in our mouse model no expression of the only identified CCL2 receptor CCR2 was observed in bronchial epithelium.
In conclusion, we have shown a rapid upregulation of L-CCR mRNA expression in the lung in a murine model of OVA-induced airway inflammation. Strong L-CCR mRNA expression was found in macrophages and in bronchial epithelium. It is conceivable that L-CCR is a functional chemokine receptor, possibly with CCL2 as a ligand. This receptor may play a role in attraction and activation of macrophages during the early phase of the inflammatory response in the lung.
Footnotes
Acknowledgements
Supported by an unrestricted grant from the Groningen University Institute for Drug Exploration (GUIDE). Part of the research was supported by the Dutch Spinoza grant (D.S. Postma).
We wish to thank Pieter Klok and Nieske Brouwer for excellent technical assistance during the in vivo procedures and in situ hybridization experiments, respectively. We are grateful to Siep Noorman for his contribution in making the photographs.