
Abstract
VIP36 (36-kD vesicular integral membrane protein), originally purified from Madin-Darby canine kidney (MDCK) epithelial cells, belongs to a family of animal lectins and may act as a cargo receptor. To understand its role in secretory processes, we performed morphological analysis of the rat parotid gland. Immunoelectron microscopy provided evidence that endogenous VIP36 is localized in the trans-Golgi network, on immature granules, and on mature secretory granules in acinar cells. Double-staining immunofluorescence experiments confirmed that VIP36 and amylase co-localized in the apical regions of the acinar cells. This is the first study to demonstrate that endogenous VIP36 is involved in the post-Golgi secretory pathway, suggesting that VIP36 plays a role in trafficking and sorting of secretory and/or membrane proteins during granule formation.
N
VIP36 (36-kD vesicular integral membrane protein) was originally purified from detergent-insoluble complexes of Madin-Darby canine kidney (MDCK) epithelial cells (Fiedler et al. 1993). Sequence analysis showed that VIP36 belongs to the family of animal lectins, it is homologous with leguminous lectins, and may be conserved from yeast to mammals (Fiedler and Simons 1994,1995).
Endoplasmic reticulum-Golgi intermediate compartment (ERGIC)-53 is also a member of the lectin family and proved to be a mannose-specific lectin localized to the ERGIC (Appenzeller et al. 1999). It has been hypothesized that VIP36 and ERGIC-53, act as cargo receptors facilitating transport and sorting of protein from the ER to the plasma membrane (Fiedler et al. 1994). Overexpressed VIP36 was localized in the post-Golgi secretory pathway of MDCK cells, and this was confirmed by the findings that the plasma membrane fraction contained a significant amount of VIP36 (Fiedler et al. 1994; Fiedler and Simons 1995). However, contradictory results, i.e., partial co-localization of VIP36 and coatomer involved in trafficking along the early secretory pathway, have been reported (Füllekrug et al. 1999; Dahm et al. 2001). To explore the distribution, we produced a specific antibody against the N-terminus (which faces the luminal space) of VIP36 and developed a strategy for immunoelectron microscopic analysis of VIP36 in cells. Ultrastructural analysis of amylase-secreting acinar cells of the rat parotid gland revealed that VIP36 was localized not only in the pre-Golgi early secretory pathway but also in the post-Golgi secretory pathway from the trans-Golgi network (TGN) to the plasma membrane.
Materials and Methods
Reagents and Antibodies
All the chemical reagents used, most of which were purchased from Sigma (St Louis, MO), were of analytical grade. The anti-VIP36 polyclonal antibody immunoglobulin against the luminal tail of VIP36 was raised in rabbits in our laboratory. The specificity of the antibody used was described in detail in a previous paper, in which no other band was detectable by immunoblotting and overexpression of VIP36 caused a specific increase of a single band corresponding to a 36-kD protein in MDCK cells (Hara-Kuge et al. 2002). It should be mentioned that our VIP36 antibody has never been observed to crossreact with ERGIC-53. Two different types of anti-amylase antibody, sheep polyclonal anti-amylase serum and a mouse monoclonal antibody, were purchased from Biogenesis (Poole, UK) and QED Bioscience (San Diego, CA) respectively. The following secondary antibodies were used: goat anti-rabbit immunoglobulin (Ig) G and anti-mouse IgG coupled to FITC or Texas Red, donkey anti-sheep IgG with Texas Red (Cortex Biochem; San Leandro, CA) and goat anti-rabbit IgG conjugated with 5- or 10-nm gold particles (EY Laboratories; San Mateo, CA).
Immunoelectron Microscopy
Young male Wistar rats weighing 80–100 g were used. They were anesthetized with pentobarbital and fixed by perfusing 5% acrolein in 1/15 M phosphate buffer (PB), pH 7.4, through the aorta at 37C. Then the parotid glands were removed, cut into small pieces, which were washed well with PB, and immersed in a fixative composed of 2% paraformaldehyde, 0.125% glutaraldehyde, and 0.1% picric acid in PB overnight at 4C (Shimada et al. 1998). The tissues were postfixed in 1% osmium tetroxide and 7% sucrose in PB for 2 hr at 4C, dehydrated with a graded ethanol series at 0C, embedded in Lowicryl K4M, and cured under UV light for 3 days at — 35C. Ultrathin sections were treated with 3% hydrogen peroxide at room temperature (RT) for 10 min, washed three times with water, and then treated with 0.1% ammonium chloride for 10 min. After washing well with water, the ultrathin sections were incubated with 10% normal goat serum in PBS for 1 hr and then incubated with the anti-VIP36 antibody immunoglobulin adequately diluted with 100 mM PBS, pH 7.4, containing 1% BSA for 24 hr at 4C. After rinsing well with PBS, the sections were incubated for 1 hr with 10-nm (for parotid acinar cells) or 5-nm (for GH3 and Vero cells) colloidal gold-labeled anti-rabbit IgG (1:200) at RT, followed by rinsing with PBS and then water. Finally the sections were stained with uranyl acetate and lead citrate and examined with an electron microscope (Hitachi H-7500).
Confocal Laser Scanning Microscopy
For comparison with the electron microscopic data, we used the same fixing procedure for the tissues subjected to immunofluorescence analysis. As described above, the tissues were fixed with acrolein, washed well with PBS, then fixed with paraformaldehyde, a very small amount of glutaraldehyde and picric acid, washed well with PBS, and treated with hydrogen peroxide and ammonium chloride to quench any aldehyde remaining in the specimens. The tissues were immersed in 30% sucrose in PBS overnight at 4C, frozen on dry ice, and 6-μm-thick frozen sections were cut with a cryostat at —20C and mounted on gelatin-coated cover glass slides. The sections were permeated with PBS containing 1% Triton X-100 and 1% BSA for 1 hr, treated with 10% normal goat serum and/or donkey serum for 1 hr, and then incubated with the required appropriately diluted antibody or cocktail of antibodies [anti-VIP36 (1:200) and/or anti-amylase (1:100)] in PBS containing 1% BSA for 24 hr at 4C. After washing with PBS, the cells were incubated with the required fluorescence-labeled secondary antibody (1:100) in PBS containing 1% BSA for 1 hr at 20C. Then all the specimens were washed well and mounted in 50% glycerol in Tris-HCl buffer (pH 8.6) containing 50 mg/ml 1,4-diazabicyclo-octane (Aldrich Chemical; Milwaukee, WI), which was used as an anti-bleaching agent. The present fixation procedure completely inhibited nonspecific fluorescence in tissues. The results of the confocal laser scanning microscopy (Olympus, LSM300) and double-labeling experiments were obtained simultaneously to exclude any artifacts resulting from sequential acquisition. Both channels were adjusted to ensure that the maximal fluorescence intensity remained within the recording range.
Cell Culture
As endocrine cells, GH3 cells, which are prolactin (PRL)-and growth hormone (GH)-producing rat pituitary adenoma cells, were provided by Dr. R. Horiuchi (Gunma University School of Medicine). As epithelial cells, Vero cells (African green monkey kidney epithelial cell line) were also provided. They were grown in DMEM supplemented with sodium pyruvate (1 mM), glutamine (2 mM), non-essential amino acids (200 μM), epidermal growth factor (20 nM), 10% horse serum, 2.5% fetal calf serum, 48 mg l−1 penicillin and 100 mg l−1 streptomycin on a glass slide in 100-mm plastic culture dishes at 37C in 5% CO2.
Cells cultured on glass slides were fixed for 5 min with 5% acrolein in 1/15 M phosphate buffer (PB), pH 7.4, at 37C, washed three times with PB, then fixed with PB containing 1% paraformaldehyde, 0.125% glutaraldehyde, and 0.1% picric acid for 1 hr at 37C followed by 3 hr at 4C (Shimada et al. 1998). The cells were postfixed with 1% osmium tetroxide and 7% sucrose in PB for 1 hr at 4C, dehydrated with a graded ethanol series at 0C, embedded in Lowicryl K4M, and cured under UV light for 3 days at — 35C.
Immunoblotting Analysis
Using the method of Hara-Kuge et al. (2002), rat tissues were homogenized in 1 ml of 250 mM sucrose, 10 mM Hepes (pH 7.4), and 2 mM EGTA using a Dounce homogenizer. The post-nuclear supernatant was prepared by centrifuge of the homogenate at 600 × g for 10 min at 4C. The supernatant was further centrifuged for 1 hr at 10,000 × g. The pellet, called the membrane fraction, was lysed by incubation on ice for 30 min in lysis buffer (50 mM Tris-HCl buffer, pH 7.0, containing 1% NP-40, 150 nM NaCl, and a protease inhibitor cocktail). The lysate was centrifuged at 3000 rpm for 10 min at 4C and the resulting supernatant was referred to as the detergent extract. Then 25 μg of the protein of each tissue was separated by electrophoresis on a 15% polyacrylamide gel and transferred to nitrocellulose membranes. VIP36 protein was detected with anti-VIP36 antibody immunoglobulin followed by HRP-conjugated secondary antibody and visualized using Chemiluminescence Reagent (Amersham Pharmacia Biotech UK; Fairlawn, NJ).
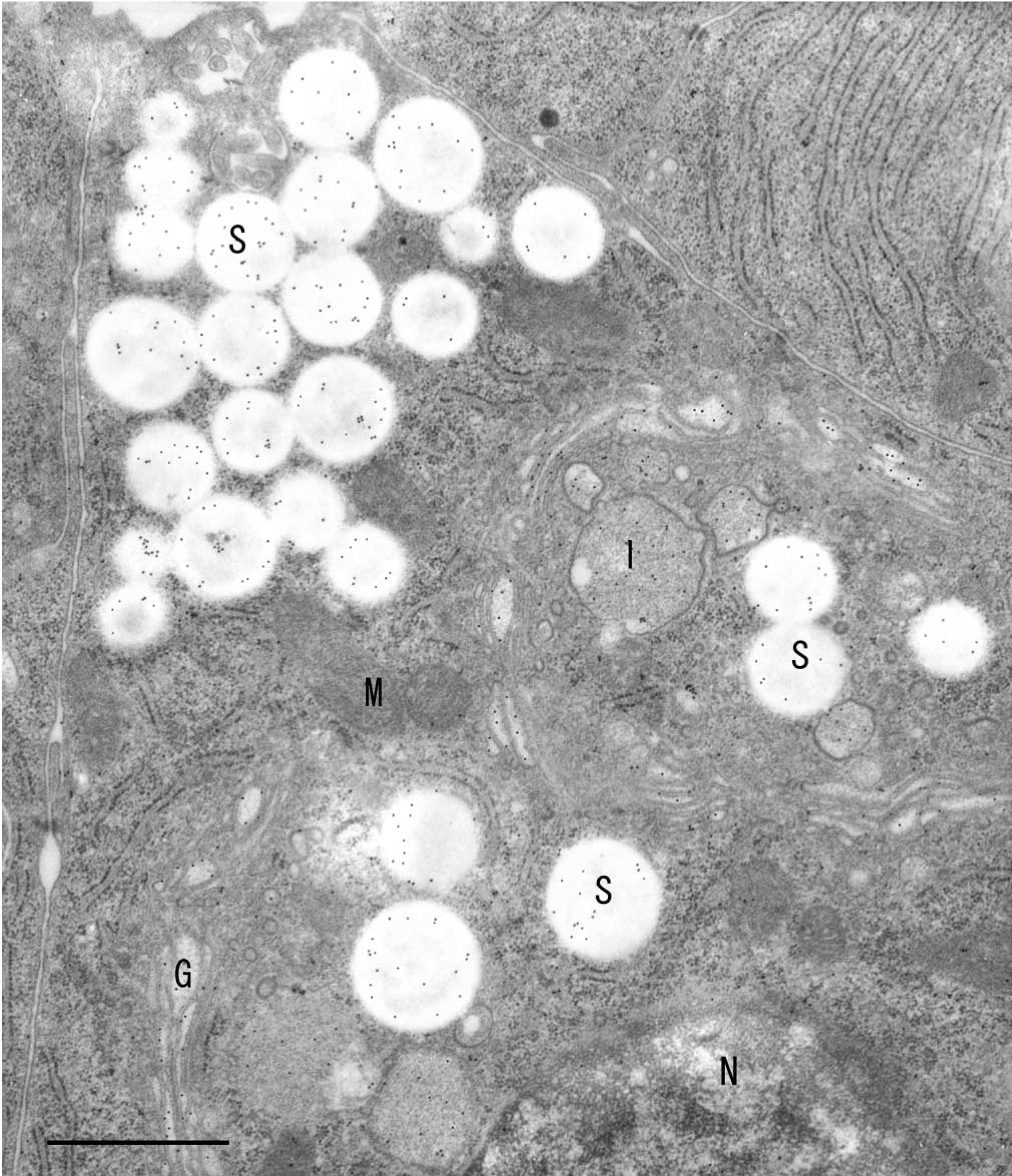
Immunoelectron microscopic localization of VIP36 in a rat parotid acinar cell. 10-nm gold particles were used. Localization of VIP36 was prominent in the Golgi apparatus (G), the immature granules (I), and the mature secretory granules (S). M, mitochondria; N, nucleus. Bar = 2 μm.
Results
The serous acinar cells of the rat parotid gland contained secretory granules that had accumulated in the subcellular portion between the nucleus and the apical cell membrane surface. The Golgi apparatus consisting of three to six cisternae, arranged in typical stacks of a cis-face to a trans-face according to polarity, was usually found in an apical, paranuclear position in the cells. Gold particles were observed on almost all the secretory and immature granules and in the neighboring trans-Golgi cisternum (Figure 1). We have shown a control section treated by preadsorption with VIP36 (Figure 2). On the control section, we observed only a few gold particles in various areas, including the nucleus, although their number was not significant. Another control section stained with preimmune serum instead of antibody immunoglobulin showed similar results. Significant numbers of gold particles were observed only on sections stained with the specific antibody immunoglobulin against VIP36. Compared to a control section (Figure 2), slight but significant numbers of gold particles were also distributed in the area of the endoplasmic reticulum.
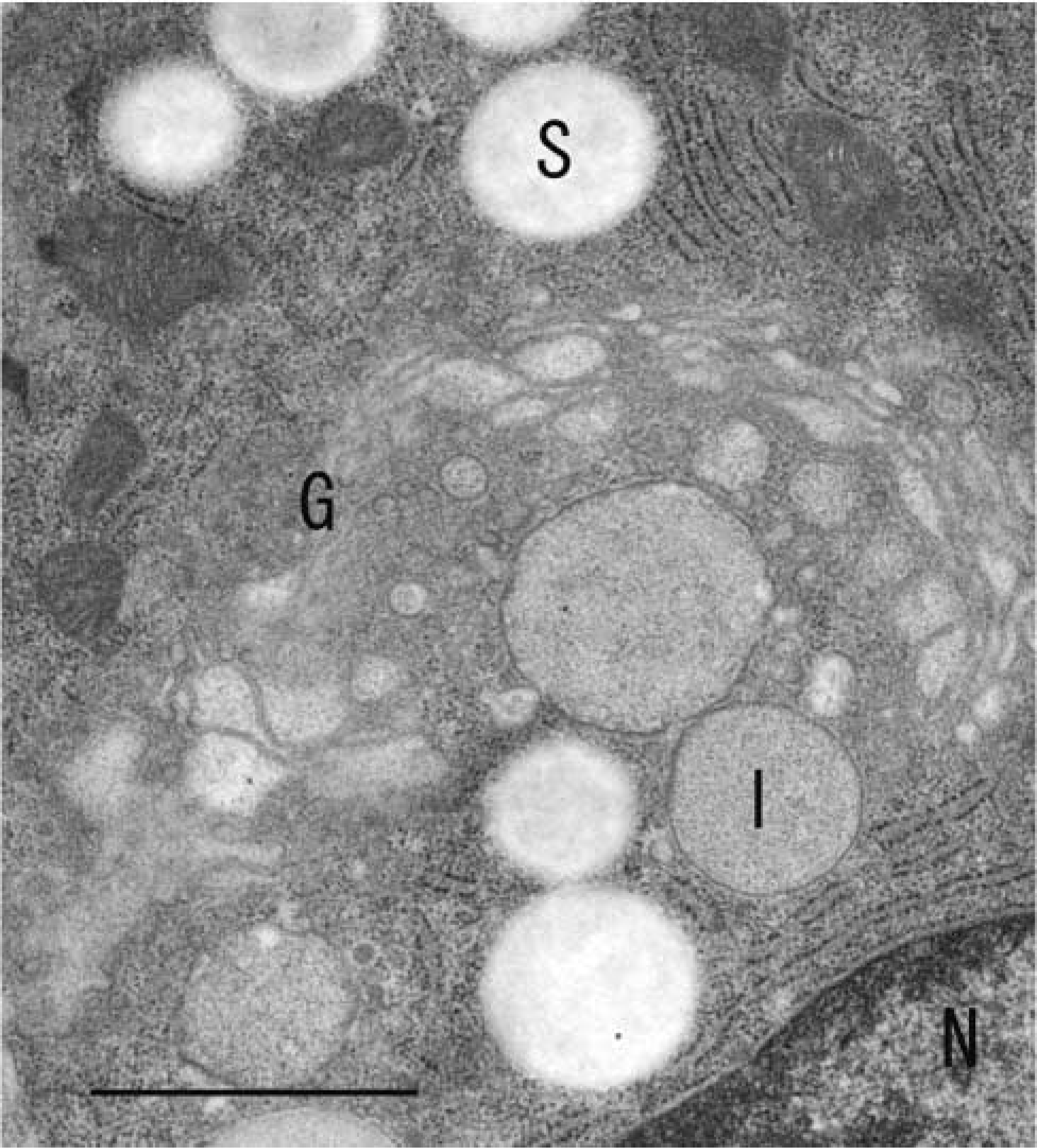
Control section stained with the antibody previously adsorbed by an excess amount of VIP36. Nonspecific distributions of the gold particles were rarely observed. G, Golgi apparatus; I, immature granules; S, mature secretory granules; N, nucleus. Bar = 2 μm.
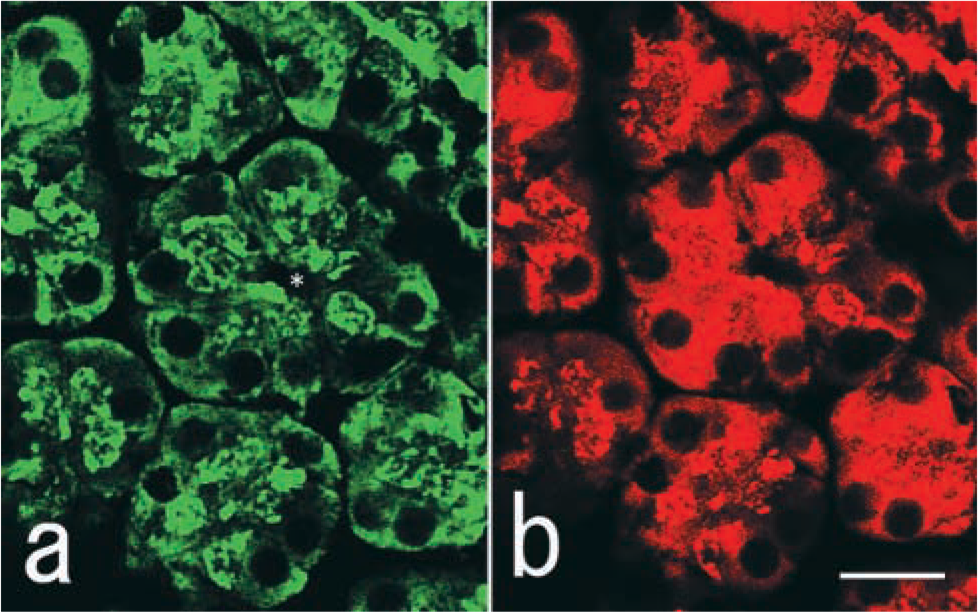
Double-staining immunofluorescence of VIP36 (a) and amylase (
Immunofluorescence microscopy showed that VIP36 was widely distributed in the apical regions of the terminal serous cells. In an attempt to investigate colocalization of secretory proteins, we compared the distributions of VIP36 and amylase. Marked but not complete overlap of VIP36 and amylase in acinar cells was observed (Figure 3a and 3b). Co-localization of VIP36 and amylase was obvious in the apical paranuclear regions of the acinar cells of the gland.
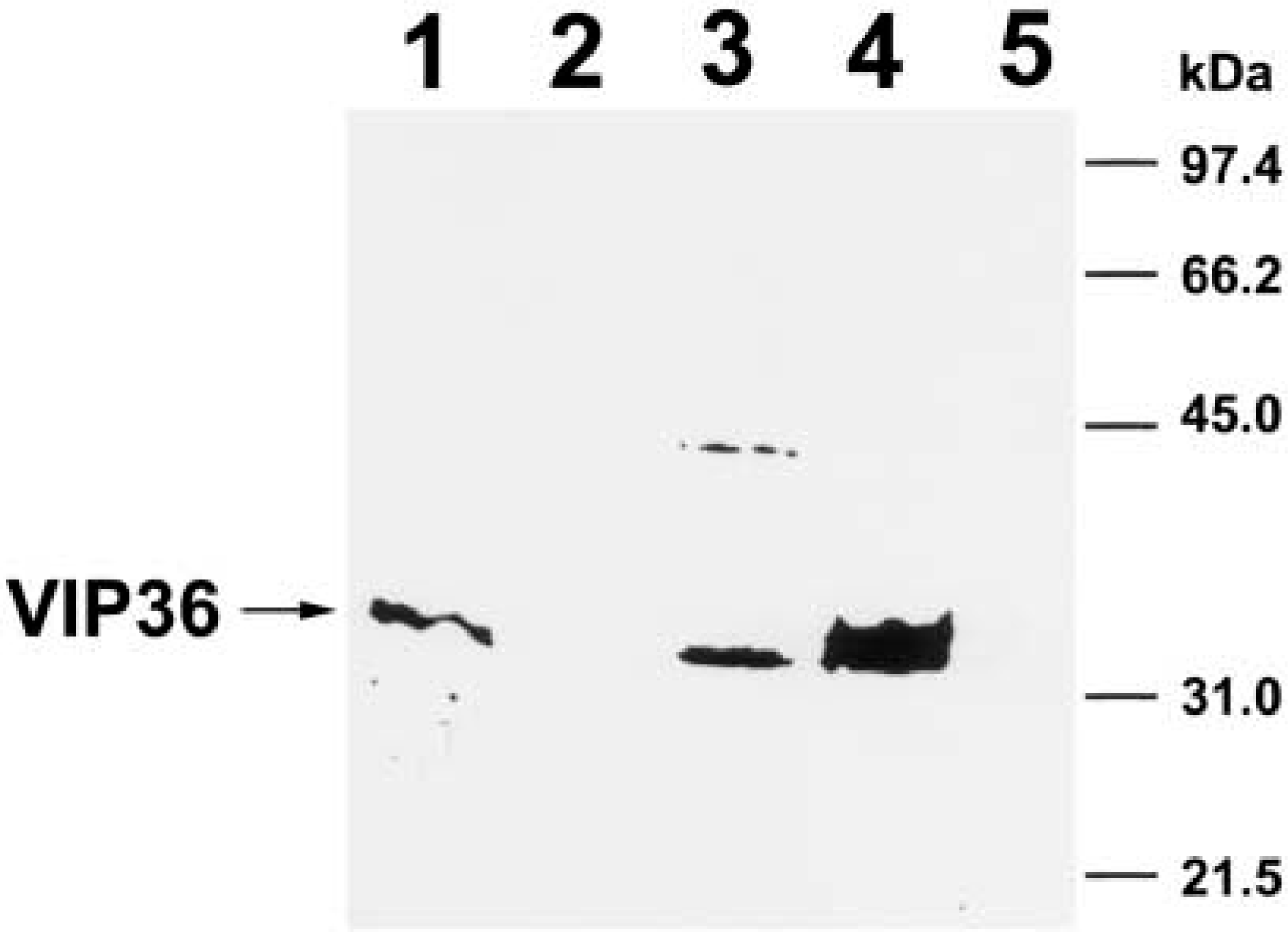
An entire gel of immunoblotting analysis in various rat tissues. 25 μg of the protein of each tissue was separated by electrophoresis on a 15% polyacrylamide gel and transferred to nitrocellulose membranes. Lane 1, adrenal cortex; Lane 2, posterior pituitary; Lane 3, submandibular gland; Lane 4, parotid gland; Lane 5, brain (cerebral cortex).
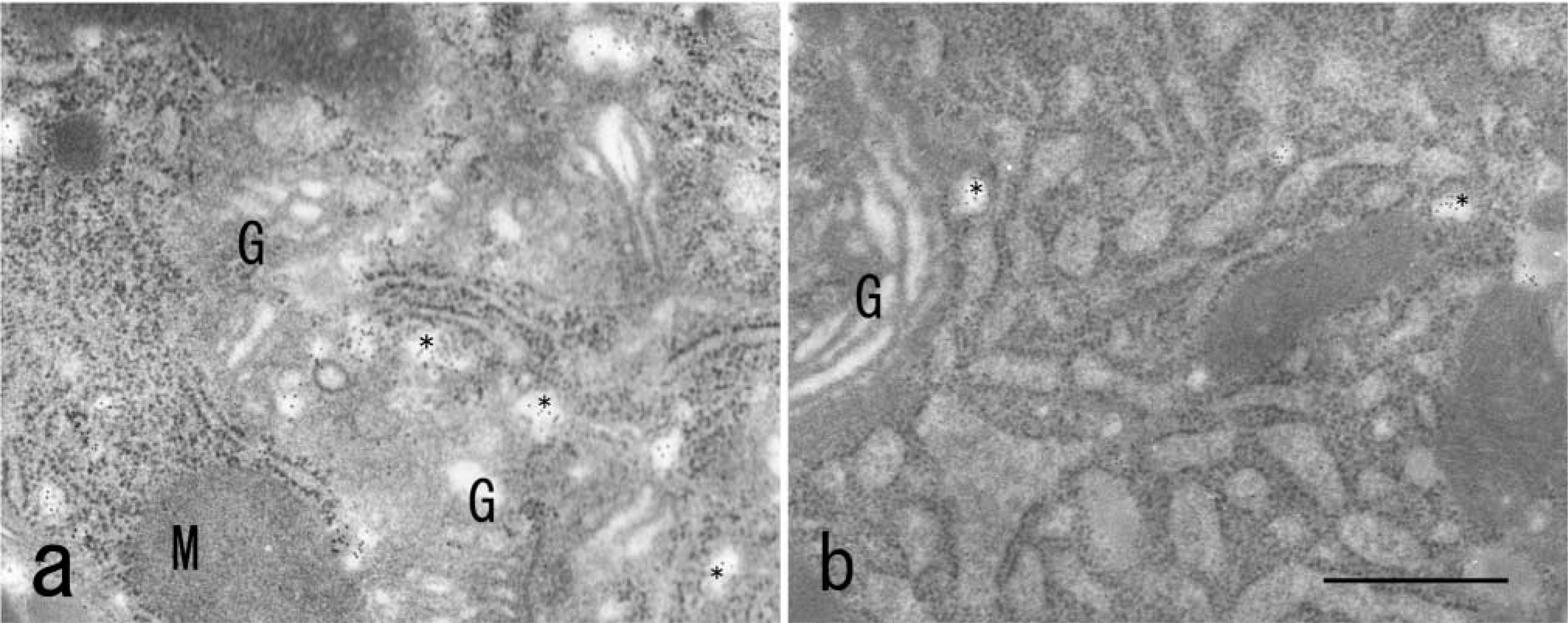
Immunoelectron microscopic localization of VIP36 in an epithelial Vero cell (
An entire gel showing immunoblot analysis of VIP36 in the parotid gland tissue yielded a strong immunopositive single band with a molecular mass of about 36 kD (Figure 4). In the parotid gland, no other band could be detected except for a single band corresponding to a 36-kD protein (Figure 4).
Immunoelectron microscopy of endocrine GH3 cells and epithelial Vero cells provided evidence that endogenous VIP36 is localized mainly in 70–100-nmdiameter vesicles around the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) between the endoplasmic reticulum and the neighboring cis-Golgi cisternum (Figure 5a and 5b).
Although we prepared two VIP36 antibody immunogobulins from two different rabbits, we obtained almost all the same results from the two antibodies. Two different antibodies against amylase also showed the same results.
Discussion
Recently, intracellular lectins were demonstrated to play important roles in vesicular transport (Hauri et al. 2000; Majoul et al. 2001). They may work as receptors (mannose-6-phosphate receptors) that recognize markers for lysosomal enzymes, as molecular chaperones (calnexin and calreticulin), and as transport cargo receptors (ERGIC-53 and Emp47p) (Ou et al. 1993; Nauseef et al. 1995; Schröder et al. 1995; Appenzeller et al. 1999). ERGIC-53 is an ERGIC marker and is identical to MR60, which is a mannosespecific membrane lectin with a carbohydrate-binding domain homologous with those of leguminous plant lectins (Fiedler and Simons 1994). The N-terminal luminal domain of ERGIC-53, which includes the carbohydrate-binding domain, has an amino acid sequence very similar to that of VIP36. VIP36 was originally isolated from the detergent-insoluble, glycolipid-enriched complexes containing apical marker proteins of MDCK cells (Fiedler et al. 1993). The above results imply that VIP36 is an intracellular lectin involved in the intracellular transport of some glycoproteins (Fiedler and Simons 1994,1995; Itin et al. 1995).
Inconsistent with parotid acinar cells, Füllekrug et al. (1999) detected endogenous VIP36 only in the pre-Golgi secretory pathway of MDCK cells, although overexpressed VIP36 was frequently distributed both in the post-Golgi pathway and on the plasma membrane (Fiedler and Simons 1995). The discrepancy might be attributable to the different cell types studied and/or different cell functions. In cells such as exocrine cells that have polarized plasma membranes, both VIP36- and VIP36-recognized proteins were distributed predominantly apically (Fiedler and Simons 1995). In MDCK cells, overproduction of VIP36 induced a marked enhancement of the amount of VIP36-recognized glycoprotein distributed apically and stimulated apical secretion of this glycoprotein (Fiedler et al. 1994; Hara-Kuge et al. 1999; Yamashita et al. 1999). In a previous study, we showed that an apical secretory glycoprotein, clusterin, carried at least one high-mannose type glycan that was recognized by VIP36 and the rate of clusterin transport was increased by the overproduction of VIP36 in MDCK cells (Yamashita et al. 1999). Taken together, these findings and the results we obtained with parotid exocrine cells in the present study suggest that VIP36 could be involved in the transport and sorting of glycoproteins through the lectin activity in polarized secretory cells.
In a preliminary experiment, we examined the localization of VIP36 in various cells. In some endocrine cells and many exocrine cells, including submandibular glandular and pancreatic amylase-secreting acinar cells, large and small intestinal goblet cells and lacrimal and thyroid glandular secretory cells, VIP36 immunoreactivity was detected along the post-Golgi secretory pathways also. However, in many endocrine and some epithelial cells, such as GH3 cells, Vero cells, and MDCK cells, the distribution of VIP36 was prominent in the pre-Golgi early secretory pathway. These data suggest that VIP36 is involved in cellular vesicular transport not only from the endoplasmic reticulum to the Golgi apparatus but also from the TGN to the plasma membrane. To elucidate the function(s) of VIP36, further investigations on a wide variety of cells are needed.
Consistent with the view that the lectin activity of VIP36 is involved in the transport and sorting of glycoproteins, several studies have indicated that N-linked glycan is one of the sorting signals for apical transport (Scheiffele et al. 1995; Füllekrug et al. 1999; Yamashita et al. 1999). It is well known that VIP36 has one potential N-linked glycosylation site (Fiedler and Simons 1995). Erythropoietin, an apical secretory protein, was secreted from both the apical and basolateral domains of the plasma membrane when its N-glycosylation site was removed by site-directed mutagenesis (Kitagawa et al. 1994). Similarly, Scheiffele et al. (1995) showed that growth hormone, which is a non-glycosylated protein secreted both from the apical and basolateral domains of the plasma membrane, was secreted preferentially from the apical membrane when an N-glycosylation site was introduced by mutagenesis.
In parotid acinar cells, localization of VIP36 was not detected on the plasma membrane in the present experiment. The distribution appeared to contradict the previous data for MDCK cells but could not rule out the possible distribution of VIP36 on the plasma membrane. The failure to find any distribution might have been due to the present fixation method, in which animals were perfused with saline and then subjected to chemical fixation. Unlike the case for separate cells, long-term fixation is needed for rats, and we considered that the changes in distribution might have occurred particularly in the surface plasma membrane. Our present chemical fixation method could not resolve the question of whether the VIP36 was recycled or exocytosed. Rapid freeze-fixation might resolve this problem in the future.
Comparing to endocrine GH3 cells and epithelial Vero cells, the parotid acinar cells showed a remarkable distribution following three portions: (a) the Golgi apparatus, (b) immature granules, and (c) mature secretory granules. This raises another question: what cargo protein(s) does the lectin VIP36 carry in the parotid acinar cells? Zymogen granules contain amylase. These is a possibility that membrane fractions containing VIP36 were dispersed and deposited in the secretory granules with cargo protein(s). Alpha-amylase contains glycosylation sequences and glycosylation modification might occur during the processing of a multitude of precursor proteins (Rindler et al. 2001). In addition to amylase, the zymogen granules in the acinar cells contain a wide variety of secretory proteins and membrane proteins, such as muclin (De Lisle and Ziemer 2000), heat shock protein (Lee et al. 2000), syncollin (Antonin et al. 2002), ZG-46p (Chen et al. 1997), ZG-29p (Kleene et al. 2000), chaperonin 60 (Le Gall and Bendayan 1996), GP2 (Lowe et al. 1994), Rab3 protein (Valentijin et al. 1996; Raffaniello et al. 1999), mucin (Kinoshita et al. 1999), epidermal growth factor (Pascall and Brown 1997; Lohinai et al. 1999), VAMP-2 (Fujita-Yoshigaki et al. 1996) and others, all of which may play different roles during secretory processes. Follow-up experiments are required to identify the cargo protein(s).
Footnotes
Acknowledgements
We thank Drs S. Oono and K. Hori (Yamanashi University School of Medicine), respectively, for valuable suggestions and letting us carry out confocal laser scanning microscopy freely.