
Abstract
Differentiating apoptosis from necrosis is a challenge in single cells and in parenchymal tissues. The techniques available, including in situ TUNEL (
Keywords
I
To overcome these difficulties, a new technique called comet assay, or single-cell electrophoresis, for detection of apoptosis has been described (Olive et al. 1993; Godard et al. 1999). This assay can detect various forms of DNA strand breakage dependent on the pH of electrophoresis (Collins 2002). Under alkaline conditions (pH >13), it detects single-strand breakage, double-strand breakage, excision repair site, and alkaline-labile sites (Abt et al. 1997). Under neutral conditions, it mainly detects double-strand DNA breakage (Olive et al. 1991) and is therefore considered to be suitable for detection of apoptosis. Despite the need for more studies to confirm the relationship of the experimental condition to the sensitivity and specificity of the comet assay, the theoretical advantages of the comet assay for the detection of apoptosis are as follows: (a) it has higher sensitivity than the ladder assay (Barbouti et al. 2002) and TUNEL staining (Godard et al. 1999); (b) it can provide more specific information about the extent and heterogenity of DNA damage compared to TUNEL staining (Olive and Banath 1995; Kindzelskii and Petty 2002); and (c) it is more accessible and feasible than EM (Collins 2002). Although the comet assay is well suited for samples from cultured cells because nuclei have to be isolated initially, it can sometimes be arduous with parenchymal tissues, such as neurons or muscles.
Clinically important, well-established models of apoptosis are those after burn injury in skeletal muscle (Yasuhara et al. 1999,2001) and ischemia/reperfusion insult to cardiac muscle (Lee et al. 2000; Matsui et al. 2001). In the present study, using these two established models, we tested the utility of the neutral comet assay.
Materials and Methods
Cell Culture
To verify the comet assay in an established system, Jurkat cells were used as a first step. Jurkat cells (clone E6.1) were provided by Dr. Junying Yuan at Harvard Medical School. The cells were grown in suspension in RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin, and streptomycin using a standard cell culture procedure.
Induction of Apoptosis and Necrosis in Jurkat Cells
Jurkat cells were seeded in flat-bottom plates at an initial density of 2 × 105 cells/ml and cultured for 4, 8, 13, and 24 hr in the above medium in the presence or absence of the following reagents. For induction of typical apoptosis, the cells were cultured with 100 ng/ml anti-Fas antibody (clone CH-11; Upstate Biotechnology, Lake Placid, NY) or 1 μM staurosporin. For induction of typical necrosis, the cells were cultured with 25 μM N-ethylmaleimide (NEM) or were heat-killed at 65C for 30 min. As controls, the cells were cultured with 100 ng/ml of the isotype-matched non-immune mouse IgM or the equivalent concentration of respective vehicle [ethanol for NEM and dimethyl sulfoxide (DMSO) for staurosporin].
Cytotoxicity Assays and Flow Cytometric Staining
Cell death was quantified by flow cytometry as previously described (Aubry et al. 1999). Briefly, cells were washed in 4C PBS, pelleted, and resuspended in 0.5 ml of hypotonic fluorochrome solution containing 50 μg/ml propidium iodide (PI) (Sigma; St Louis, MO), 0.1% sodium citrate, and 0.1% Triton X-100 (Sigma) to quantitate the cellular DNA content under the permeabilized condition. Phosphatidyl-serine (PS) exposure due to flipping of the plasma membrane, a concomitant feature during apoptosis, was evaluated by annexin V–FITC staining. Cells were washed with PBS and incubated in a solution of 0.5 μg/ml FITC-labeled annexin V (Roch; Nutley, NJ). At the same time, cells were stained by the PI exclusion method for the detection of all the dead cells. Cells were then analyzed by flow cytometry.
Burn/Sham Burn Procedure in Rat for Induction of Skeletal Muscle Apoptosis
The protocol for the studies was approved by the institutional Animal Care Committee. Adult male Sprague–Dawley weighing 200 g were purchased from Taconic Farms (Germantown, NY). The rats were anesthetized with sodium pentobarbital (50 mg/kg bw) administered IP, and were divided into burn and control groups. Rats in the burn group received thermal injury to 40% of total body surface area on the trunk and the back according to the protocol previously described (Ikezu et al. 1997; Yasuhara et al. 2001). Briefly, they were immersed in 80C water for 15 sec on the back and both flanks and for 8 sec on the abdominal side. This procedure, confirmed by microscopy, does not cause direct burn injury to deeper muscle tissue (Yasuhara et al. 1999,2001). Fluid resuscitation was performed by injecting 10 ml of normal saline. Animals in the control group were given sham burns by immersion in lukewarm water at room temperature. All other procedures were conducted exactly the same for both groups of rats. At the scheduled time point (12 hr, 1 day, or 3 days) the animals were sacrificed and the rectus abdominis muscle tissues excised immediately for analysis.
Ischemia/Reperfusion to Rat Model of Cardiac Muscle Injury
The procedure for ischemia/reperfusion injury has been described previously (Matsui et al. 2001). Briefly, adult male Sprague–Dawley rats weighing 280 g were anesthetized with pentobarbital, intubated, and ventilated (SAR-830; CWE, Ardmore, PA). After thoracotomy, the left anterior descending coronary artery (LAD) was ligated at 4 mm from its origin. Five minutes after ischemia, 200 μl of fluorescent microspheres (10-μm FluoSpheres; Molecular Probes, Eugene, OR) was injected into the left ventricular cavity. After 30 min, the LAD ligature was released and reperfusion was visually confirmed. For sham ischemia/reperfusion injury, thoracotomy was performed without LAD ligation. At 24 hr after operation, rats were sacrificed and hearts were dissected out. The harvested ischemia/reperfused hearts were cut into two parts (i.e., intact non-ischemic part and ischemic part) under brief exposure to a UV lamp. The ischemic part lacked fluorescence signal from injected microspheres, whereas the intact part gave red fluorescence from the perfused microspheres.
Isolation of Nuclei Using Percoll Density Gradients
The skeletal and cardiac muscle tissues from the sacrificed rats were quickly placed in ice-cold PBS. Skeletal muscle and ventricular heart muscle were trimmed to remove bulk connective tissue and minced with scissors. Nuclei were prepared by a modification of the procedure described by Hahn and Covault (1990) and Kuehl (1977). One gram of trimmed muscle was homogenized in 25 ml of Buffer A [0.3 M sucrose, 60 mM KCl, 0.15 mM spermine, 0.5 mM spermidine, 0.5 mM ethylene glycol bis β-aminoethylether N,N'-tetraacetic acid (EGTA), 2 mM ethylenedinitrilo tetraacetic acid (EDTA), 14 mM β-mercaptoethanol, 10 mg/ml BSA, 15 mM 4-(2-hydroxyethyl)-1-piperazine ethane-sulfonic acid (HEPES), pH 7.5) using a Polytron Homogenizer (10 mm shaft generator; Brinkmann, Westbury, NY) for 30 sec each at 60% power. The homogenate was centrifuged in a Beckman JA-20 rotor for 5 min at 3000 rpm and the pellet was re-homogenized in 20 ml of Buffer B (as Buffer A, but with 0.1 mM EGTA and 0.1 mM EDTA) for 15 sec at a setting of 70% power. Triton X-100 was added to a final concentration of 0.5% (v/v) and the sample was hand homogenized using a Teflon pestle Potter–Elvehjem tissue grinder. The resulting homogenate was filtered through 100-μm diameter nylon mesh to remove poorly disrupted tissue pieces. Percoll (Pharmacia; Piscataway, NJ) in Buffer B was added to the filtrate to a final concentration of 27% (v/v) and the mixture was centrifuged at 27,000 × g for 15 min. The nuclear layer near the bottom of the test tube was removed with a sialinized Pasteur pipette, diluted with 10 volumes of Buffer B, layered on a 1-ml pad of nuclei storage buffer [50% glycerol, 75 mM NaCl, 5 mM magnesium acetate, 0.85 mM dithiothreitol (DTT), 0.125 mM phenylmethylsulfonyl fluoride (PMSF), 20 mM Tris-HCl, pH 7.9], and centrifuged at 1000 × g for 10 min. The nuclear pellet was re-suspended in the storage buffer and was stored at −70C until the time of use.
Western Blotting Analysis
The proteins before and after separation into cytosolic and nuclear fractions were loaded on 13% polyacrylamide gels and electrophoresed. After transfer to nitrocellulose membranes, the membranes were blocked with 5% non-fat milk in TBS-T (20 mM Tris-HCl, 500 mM NaCl, pH 7.5, and Tween 0.1%) and incubated overnight at 4C with a mouse anti-myosin ventricular heavy chain antibody (Chemicon; Temecula, CA), a mouse anti-histone antibody (Chemicon), or a mouse anti-myosin heavy chain (fast-twitch fiber) antibody. Goat anti-mouse IgG (Calbiochem; La Jolla, CA) was used as second antibody. Membranes were washed with TBS-T and incubated in enhanced chemiluminescence detection reagents (Amersham International; Amersham, UK) to visualize the proteins of interest.
In Situ TdT-mediated dUTP-X Nick End-labeling Analysis
TUNEL staining was performed according to the manufacturer's instructions (Roche). Briefly, frozen tissues were cryosectioned at 7-μm thickness with the Jung Firgocut 2800E (Leica; Buffalo, NY) and fixed on slides with 4% paraformaldehyde for 10 min at RT. Samples were subjected to the reaction with terminal deoxynucleotide transferase in the presence of digoxigenin-conjugated nucleotide substrate for 30 min at 37C. After the reaction was stopped, the slides were incubated with anti-digoxigenin antibody that had been conjugated with fluorescein. Samples were counter-stained with 4',6-diamidino-2-phenylindole (DAPI).
Neutral Comet Assay
For detection of DNA fragmentation associated with apoptosis, a reagent kit (Trevigen; Gaithersburg, MD) was used according to the manufacturer's instructions. Briefly, the Jurkat cells or isolated nuclei from cardiac or skeletal muscle at a concentration of 1 × 105/ml were combined with low temperature melting agarose at a ratio of 1 to 10 volume (v/v) and spread on a slide glass. Slides were submerged in pre-cooled lysis solution (2.5 M NaCl, 100 mM EDTA, pH 10, 10 mM Tris base, 1% sodium lauryl sarcosinate, and 1% Triton X-100) at 4C for 30 min. After lysis and rinsing, slides were equilibrated in TBE solution (40 mM Tris/boric acid, 2 mM EDTA, pH 8.3), electrophoresed at 1.0 V/cm for 20 min, and then stained for 30 min in SYBR Gold. For scoring the comet pattern, 400 nuclei from each slide were counted.
Scoring system for comet assay
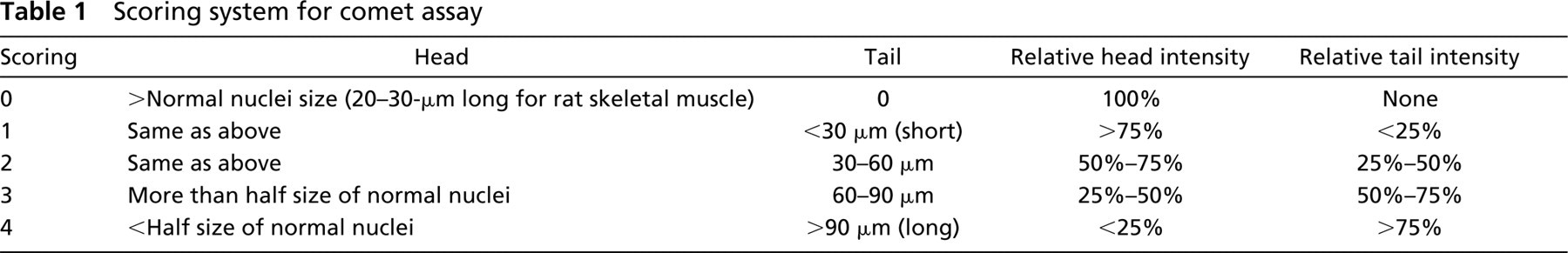
Scoring of Each Comet
For ranking each comet, we followed the original method developed by Collins (2002). The common method for scoring the comet, other than by computers, is by measuring tail length, head size, tail intensity and head intensity, (Collins 2002; Olive 2002). Table 1 shows the detailed categorization method that we used for our samples. In our system, four measurements were recorded, including the actual head size, tail length, relative head staining intensity, and relative tail staining intensity. When all of the four categories were satisfied, the matching score was given to the comet. Once any of the four categories was missed, a lower rank of score was given.
Electron Microscopy
Jurkat cell samples were harvested by low-speed centrifugation (200 × g). All the following procedures were performed at RT and the centrifugations, at each step to change the solutions, did not exceed the speed of 200 × g. After washing twice with PBS, they were fixed in iso-osmotic glutaraldehyde fixation buffer for 2 hr at RT (1.4% glutaraldehyde, 64 mM cacodylate buffer, pH 7.3). After washing three times with iso-osmotic cacodylate buffer (80 mM cacodylate buffer, 130 mM sucrose, pH 7.3), the cells were postfixed with iso-osmotic osmium tetraoxide solution (1.72% osmium, 86 mM cacodylate buffer, pH 7.3) for 1 hr. The cells were then washed three times with double-distilled water, and stained with 2% uranyl acetate in 50% ethanol solution for 2 hr. Sequential dilution series of ethanol (50%–100%) were used to dehydrate the sample. Finally, the solution was changed to 100% acetone followed by a mixture of acetone plus embedding resin (1:1) and 100% resin (Embed812: Araldyte 502:dodenyl succinic anhydride:DMP-30 = 25:15:55:1.7). After sectioning the sample at 50 nm, the sections were briefly stained with lead citrate (2–5min). Transmission electron microscopic analysis was performed with a Philips 410 at the acceleration voltage of 60 kV.
Statistical Analyses
For comparison of comet scores, the χ2 test was utilized; p<0.05 was considered statistically significant.
Results
Jurkat Cell Apoptosis vs Necrosis
Electron Microscopic Analysis. To verify our technique for neutral comet assay, Jurkat cells were induced by various stimuli to undergo apoptosis or necrosis. Fas ligation (Itoh et al. 1991) and staurosporin are known to induce apoptosis, while extensive heat treatment and NEM induce non-apoptotic (necrotic) cell death (Matteucci et al. 2000; Lecoeur et al. 2001). The normal control cells were treated with nonimmune IgM or vehicle only (ethanol and DMSO). At 8 hr after stimulation, the apoptotic group of cells (Fas or staurosporin) showed typical apoptotic morphology by EM, characterized by a rather intact organelle structure including mitochondria and by homogeneous condensation of chromatin to one side or the periphery of the nuclei (Figures 1b and 1c, arrows). The inner matrix of some mitochondria at this stage showed increased electron density as is typically observed with apoptotic cells (Figures 1b and 1c, arrowheads). The apoptotic cells at later stages (13 hr and 24 hr; data not shown) showed membrane blebbing and apoptotic body formation with fragmented nuclei, all hallmarks of typical apoptotic cells. The necrotic group of cells (NEM or heat) showed a typical necrotic pattern, with ruptured plasma membrane, irregular chromatin destruction, poorly stained cytoplasm, vacuole formation, and disrupted organelles at 8 hr (Figures 1d and 1e), and 24 hr (data not shown). These ultrastructural changes were unique in apoptotic cells and necrotic cells, respectively, and were absent in control cells (Figure 1a), which showed intact membranes and intact morphology of organelles.
Flow Cytometric Analysis. When analyzed by flow cytometry using annexin-V and propidium iodide (PI) staining at 8 hr after stimulation, the Jurkat cells treated with Fas ligation and staurosporin showed a typical apoptotic pattern, evidenced as annexin-V-positive and
Neutral Comet Assay. From morphological EM observation, the difference between apoptosis and necrosis was most prominent in Jurkat cells between 4 and 24 hr. Therefore, our focus on the comet assay for Jurkat cells was on this time window. When Jurkat cells stimulated for 13 hr were electrophoresed under neutral condition (pH 8.3) for 10 min, the apoptotic groups (Fas ligation or staurosporin treatment) showed more frequent and longer comet tails with small comet heads (Figures 3B and 3C), whereas control or necrotic groups gave distinctively less comet tail (Figures 3A, 3D, and 3E). Comet tail was scored according to Table 1. The comet patterns for the apoptotic and necrotic groups, expressed as a percentage of total cells, are shown in the histogram in Figure 3F. Cells induced to undergo apoptosis by Fas and staurosporin gave a higher score with comet assay, with more apoptotic cells categorized into class 3 or class 4 compared to the control. NEM and heat-treated necrotic cells showed only minimal comet tails (Figure 3F). The percentages of nuclei that yielded comet scores higher than 2 were 0%, 39.9%, 63.9%, 2.0%, 0% for control, Fas, staurosporin, NEM, heat-treated groups, respectively (Figure 3F). These data on percentages of nuclei with damaged DNA are close to that of apoptosis from flow cytometry (Figure 2A, lower right quadrant; 2.36%, 19.87%, 53.82%, 1.28%, and 0.80%, respectively). Furthermore, when the comet scoring pattern was compared between control and apoptosis groups and between apoptosis and necrosis groups, the scoring pattern was statistically different between control and either of the apoptotic groups, and between each apoptotic group and each necrotic group (Figure 3F, χ2 test; ∗∗ p<0.01) There was no statistical difference between control and necrosis comet patterns.
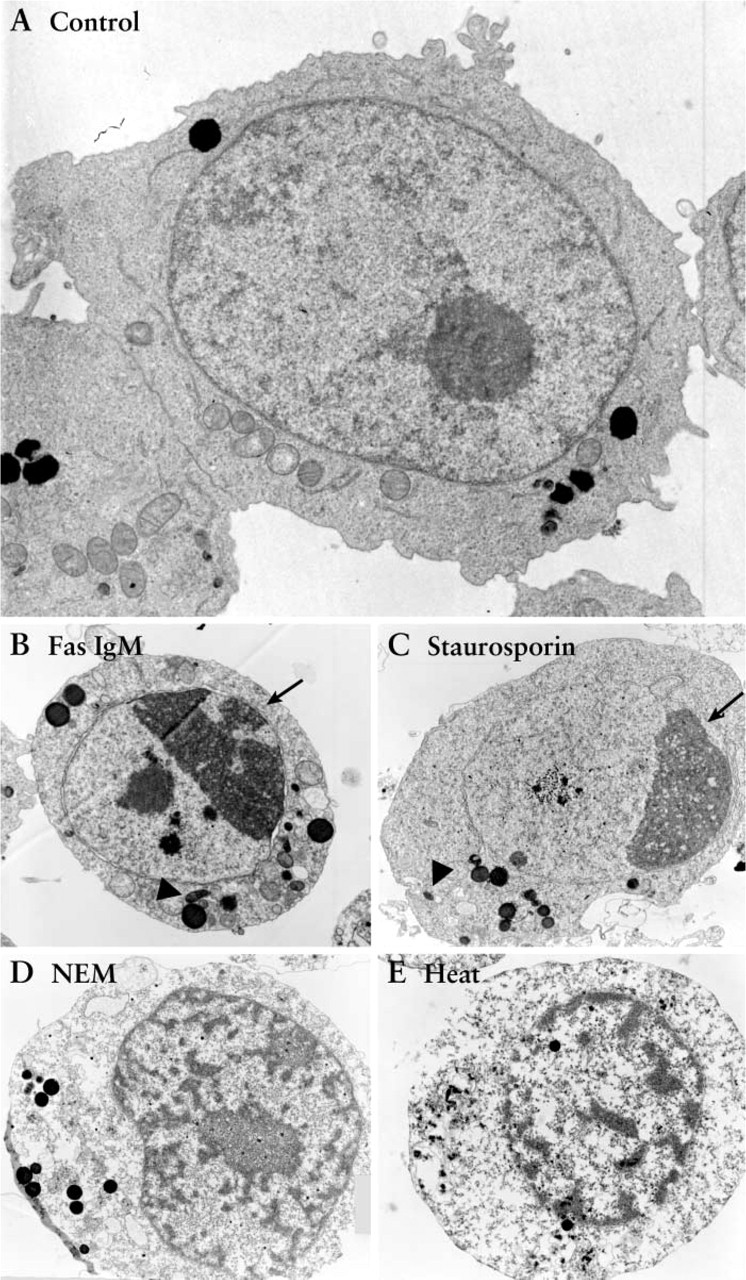
EM evaluation of apoptosis/necrosis in Jurkat cells. Jurkat cells at 8 hr after stimulation with (
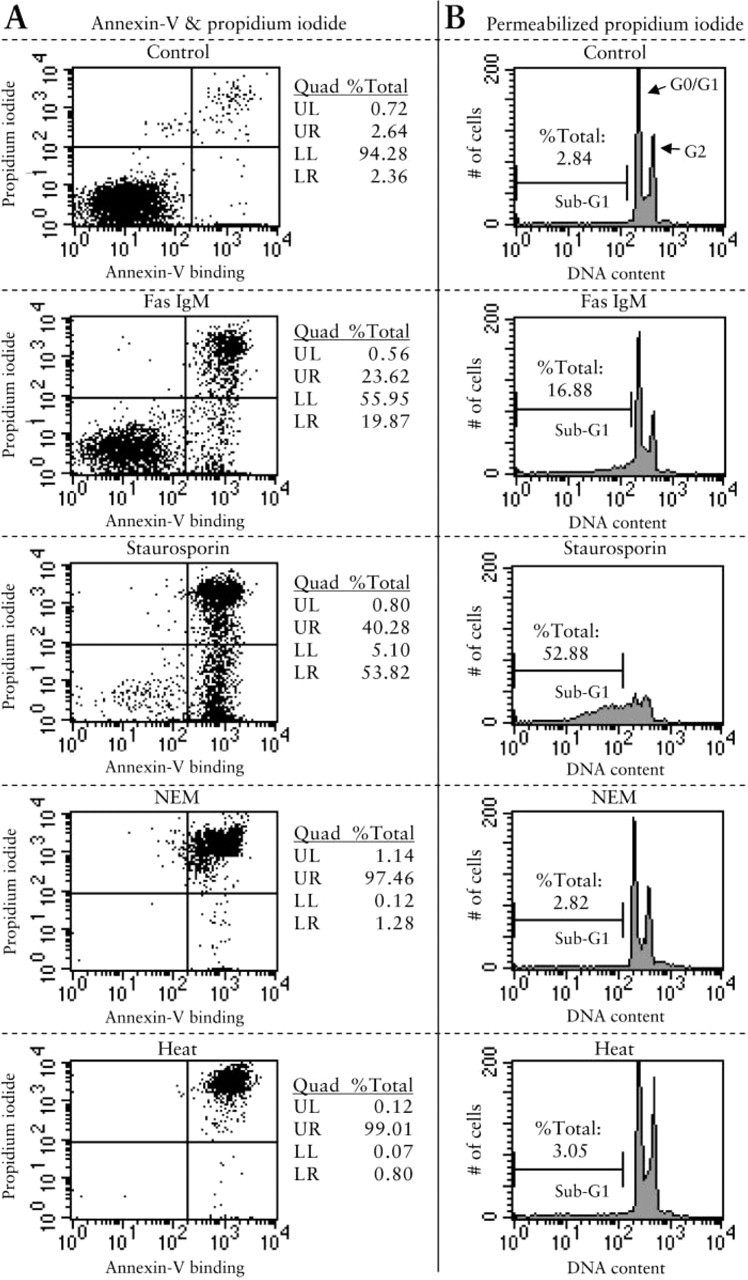
Flow cytometric analysis of Jurkat cells. Cultured Jurkat cells were exposed to Fas IgM, staurosporin, NEM, or heat. After 8 hr, the cells were harvested and stained either with (
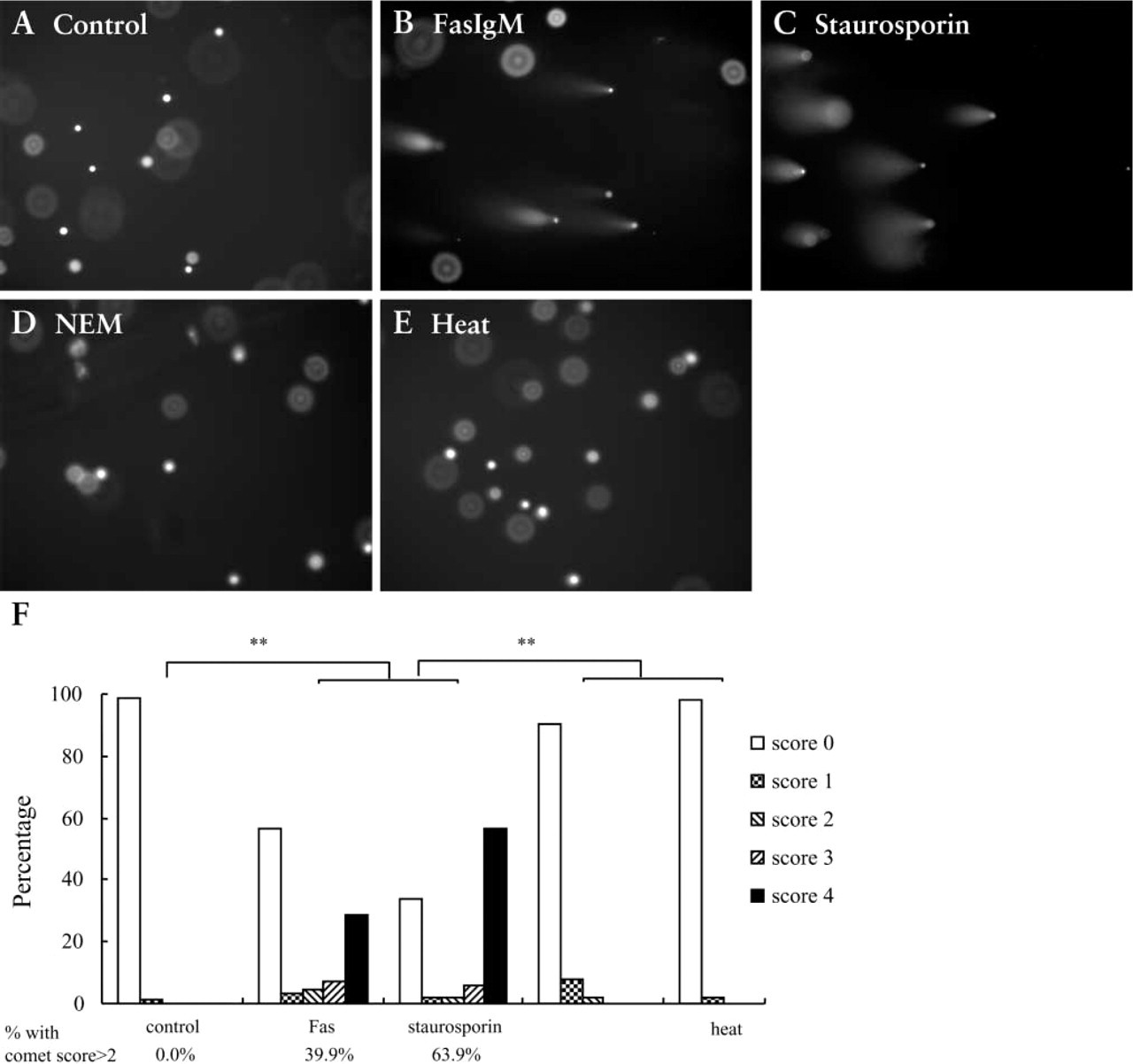
Comet assay of Jurkat cells. Cultured Jurkat cells were stained by comet assay after stimulation by different methods. Four hundred nuclei were counted for each sample. (
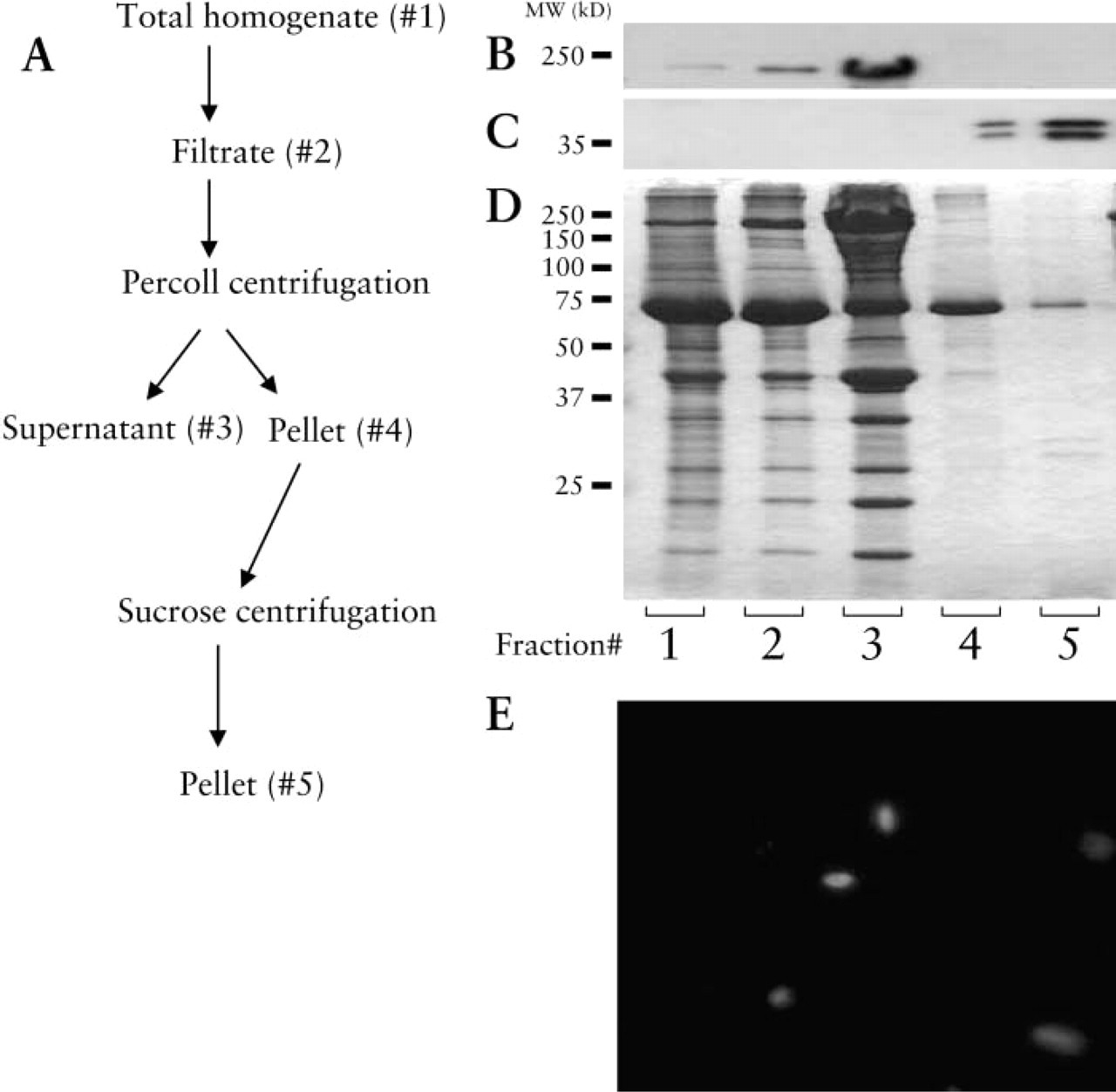
Skeletal muscle nuclei purification profile. (
Nuclear Purification Profile from Rat Skeletal Muscle
Nuclei from rat abdominal muscle, or rat heart were purified by the Percoll differential centrifugation method. To verify the purity of the isolated nuclei, we performed Western blotting of each fraction at each step during the purification procedure. Anti-histone and anti-myosin heavy chain antibodies were used to detect nuclear protein and myofibrillar protein, respectively. In the final fractions (Figure 4b, Lanes 4 and 5), there was minimal contamination by myofibrillar protein, since the band corresponding to the myosin heavy chain (200 kD) in this fraction was barely seen. At the same time, nuclei were greatly enriched in the final preparation, as shown by the histone bands (40 kD; Figure 4c, Lanes 4 and 5), compared to less-purified protein (Figure 4c, Lanes 1–3). The loss of nuclei during the purification procedure was negligible, as confirmed by lack of staining with the anti-histone antibody on the other fractions such as the supernatant (data not shown). When the shape of nuclei in controls was observed after staining with DAPI, most of the purified nuclei had a typical rod-like shape, a feature common to muscle nuclei (Figure 4e).
Apoptosis in Skeletal Muscle by In Situ TUNEL Assay
On the basis of our previous studies (Yasuhara et al. 1999,2001), muscle tissue is known to show apoptotic changes after burn injury. Under the conditions utilized, apoptosis starts from around 12 hr and peaks at day 1 and day 3 after burn injury. In situ TUNEL staining was therefore performed in cryosections of abdominal muscles obtained at 12 hr, 1, and 3 days after burn injury to rats (Figures 5A–5F). As shown in Figures 5A–5F, muscle tissue from burned rats showed TUNEL-positive nuclei, but the number of nuclei positive for TUNEL staining from sham-burned rats was minimal. In sections of the area examined, the percentages of apoptotic nuclei were as follows: 57.6%, 73.8%, 62.9% for 12 hr, 1 day, and 3 days after burn injury, respectively. Therefore, the results of the positive nuclei for apoptosis were consistent with our previous finding (Yasuhara et al. 1999) in that positive nuclei were high in tissues from rats with burns, peaking at day 1 and day 3, and low in the muscles from rats with sham burns. In addition, when DNA was extracted and run on an agarose gel, only burned animals, but not sham-burned animals, showed DNA ladder formation (data not shown).
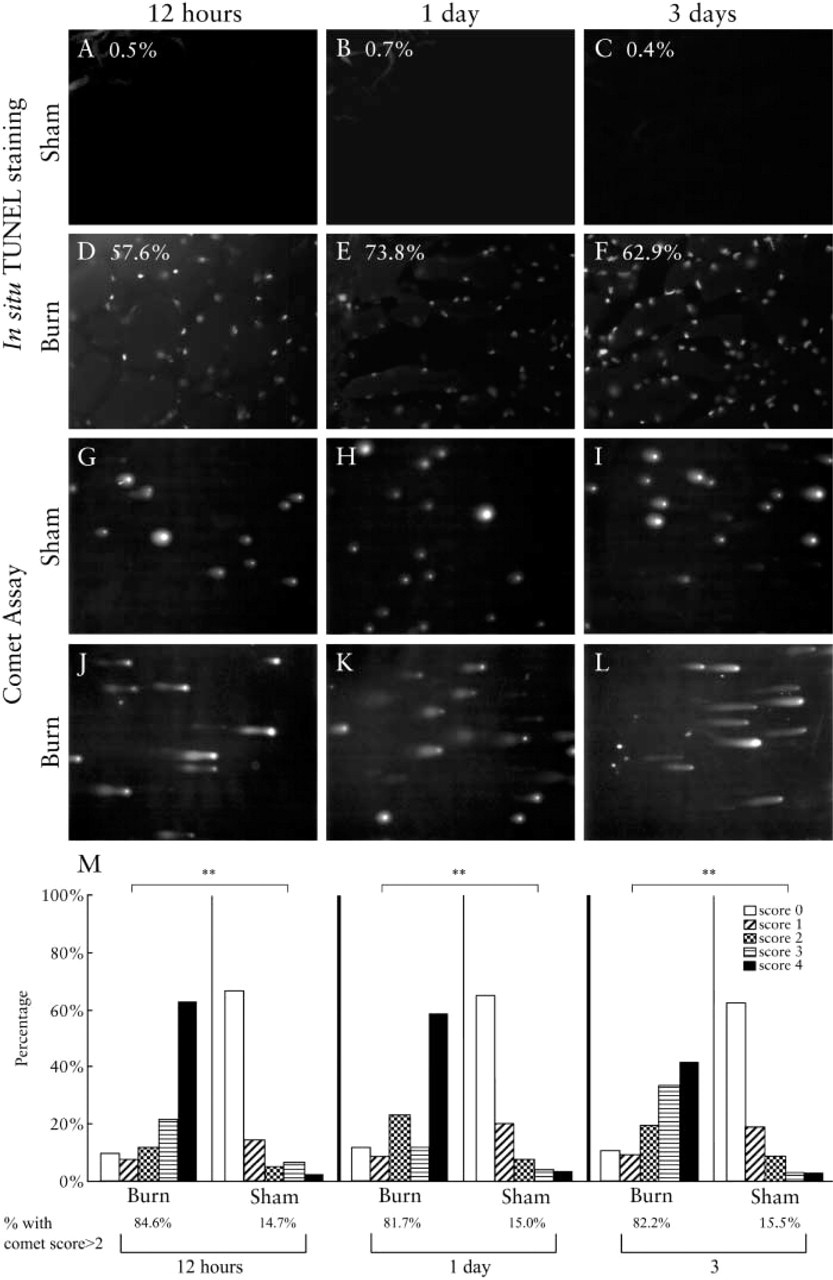
Comparison of in situ TUNEL and comet assay in abdominal muscle. (
Apoptosis in Skeletal Muscle by Comet Assay
Next, we applied the comet assay to the nuclei isolated from abdominal muscle tissues of burned and sham-burned rats. Nuclei from burned rats gave distinctively longer comet tailing (Figures 5G–5L). The samples from rats with burns had higher comet scores (class 3 and 4) than those of sham-burned rats (Figure 5M). The percentages for nuclei with scores more than 2 were 84.6% for burned, and 14.7% for shamburned tissues at 12 hr after the injury. For day 1, the ratio was 81.7% and 15.0%, respectively. For day 3, the ratio was 82.2% and 15.5%, respectively.
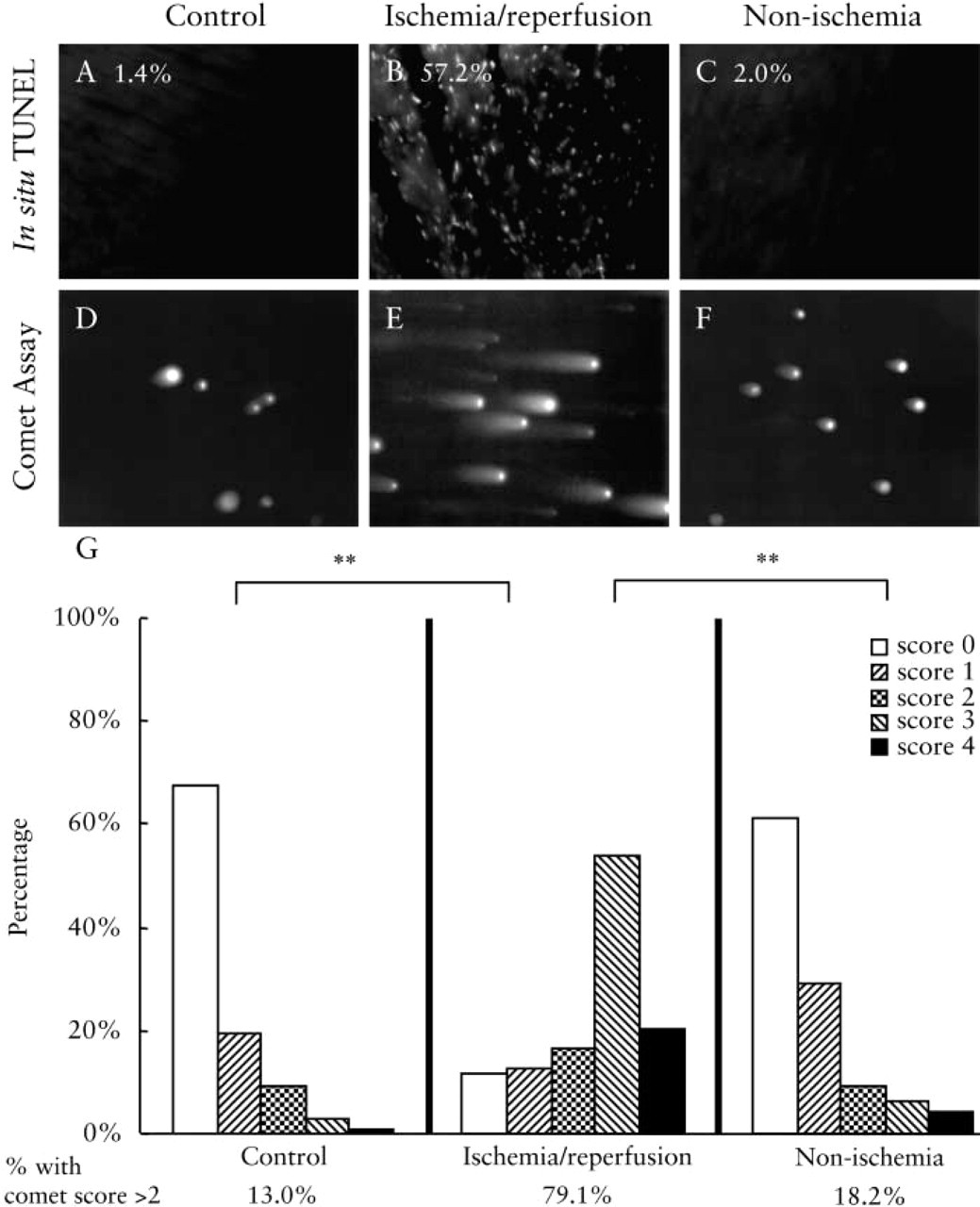
Comparison of the in situ TUNEL and comet assay in cardiac muscle. (
Apoptosis in Cardiac Muscle Demonstrated by In Situ TUNEL
Another established in vivo model of muscle apoptosis is ischemia/reperfusion injury to the heart. In the following studies, the in situ TUNEL was compared to the comet assay. As shown in Figures 6A–6C), rat cardiac muscle after ischemia/reperfusion injury exhibits TUNEL-positive nuclei (Figure 6B). When unper-turbed control heart (Figure 6A) or muscle from the intact segment of the affected heart (Figure 6C) was stained, the samples were TUNEL-negative (no staining). In sections of the area examined, the percentages of apoptotic nuclei were as follows: 1.4%, 57.2%, 2.0% for the control, ischemia, and intact parts, respectively.
Apoptosis in Rat Cardiac Muscle by Comet Assay
When the nuclei were isolated and analyzed by neutral comet assay, ischemia/reperfused rat heart segments showed distinctively higher scores of comet tailing compared to controls (Figures 6D–6F). The percentages of nuclei with scores more than 2 were 13.0%, 79.1%, and 18.2% for control, ischemia/reperfusion, and non-ischemic tissues, respectively.
Discussion
Apoptosis is not only necessary for the normal development of tissues (Wyllie et al. 1980) but is also a distinctive pathological process leading to various diseases once it is mis-regulated (Afford and Randhawa 2000). Therefore, detection of apoptosis, together with investigation of signaling pathways that mediate or inhibit apoptosis, is of importance for both scientific and medical purposes. Many methods of detection of apoptosis have been described, each with its own advantages and disadvantages. For example, in situ TUNEL or ISEL staining, the most commonly used method for detection of apoptosis, is known to have a high false-positive ratio, giving a positive signal not only with apoptosis but also with other forms of DNA damage (Sloop et al. 2001). The DNA ladder assay is highly specific for apoptotic DNA breakdown but has very low sensitivity, even if various techniques are used to refine it (Yasuhara et al. 2001). The nucleosomal ELISA method is sensitive but also can be nonspecific unless differential centrifugation method is also used to select for apoptotic DNA fragmentation. Moreover, as is the case with any other ELISA, nucleosomal ELISA depends on the sensitivity and specificity of the antibody utilized. Such assays are therefore fraught with inconsistencies, depending on the lot of the antibody used (unpublished observations). Morphological analysis by EM is a powerful method to detect apoptosis but requires skills. With studies using EM, the availability of equipment is limited. Moreover, it cannot analyze too many samples because of laborious preparation. Because only a small area can be visualized, quantification of the extent of apoptosis is also difficult by EM.
Single-cell electrophoresis, or comet assay, was originally invented more than a decade ago for the study of DNA damage of various types (Ostling and Johanson 1984). The images obtained for each cell nuclei consist of a “head” and a “tail,” the whole forming a comet-like image. Recently, the comet assay has been modified for cell culture study of apoptosis because it is easy, sensitive, and quantitative (Godard et al. 1999; Olive et al. 1993). These studies used comet assay for the detailed analysis of DNA cleavage during apoptosis in a cell culture system (Choucroun et al. 2001; Barbouti et al. 2002). In these studies, observations of early onset of apoptosis and of various types of DNA strand cleavage are demonstrated under specific conditions. The theoretical advantages of the comet assay for the detection of apoptosis are as follows: (a) it has higher sensitivity than the DNA ladder assay (Barbouti et al. 2002) and TUNEL staining (Godard et al. 1999); (b) it can provide more specific information about the extent and heterogenity of DNA damage compared to TUNEL staining (Olive and Banath 1995; Kindzelskii and Petty 2002); and (c) it is more accessible and feasible than EM (Collins 2002). However, more studies are needed to confirm the relationship of the experimental condition to the sensitivity and specificity of the comet assay.
In this study we used both typical apoptosis stimulations and necrosis stimulations to evaluate the accuracy of the comet assay. Our data show that the comet scoring pattern was distinctively different between control and apoptosis groups and between apoptosis and necrosis groups. The percentages of nuclei that yielded comet scores higher than 2 were 0%, 39.9%, 63.9%, 2.0%, 2.0% for control, Fas, staurosoprin, NEM, and heat-treated groups, respectively (Figure 3F). These comet data of percentage of nuclei with damaged DNA by comet are close to the quantitation of apoptosis by flow cytometry (Figure 2A lower right quadrant; 2.36%, 19.87%, 53.82%, 1.28%, and 0.80%, respectively). The data were consistent in that the values were high for apoptosis and low for control and necrosis groups. In our models of apoptosis and necrosis, neutral comet assay successfully differentiated the two types of cell death. Apototic nuclei showed longer comet tails with high score, whereas necrotic nuclei yielded almost no tails, which implied that this assay could be used to differentiate apoptosis from necrosis. Whereas apoptosis is defined quite well and follows certain characteristic morphological and biochemical changes, necrosis remains a rather uncharacterized process and is therefore regarded as a more heterogeneous entity. When a cell receives extreme damage, the internal cell death (apoptosis) process does not appear to function very well, and therefore shows a distinct necrotic phenotype compared to apoptosis (Lecoeur et al. 2001). During cell death from necrosis, there is a lack of proper DNA degradation processes that are usually initiated by activation of caspases, and therefore largely unfragmented DNA is left in the cell (Martin et al. 1991). Even when DNA is degraded, it appears that the degradation often gives rise to much larger fragments (MacManus et al. 1997). Therefore, it is believed that different types of DNA degradation processes are involved in apoptosis and necrosis, each using different enzymes and each having different kinetics (Hayashi et al. 1998). The neutral comet assay is presumed to have better sensitivity for double-stranded DNA breakage and less for single-strand breakage or other types of DNA damage compared to alkaline comet assays (Collins 2002). We postulate that the typical apoptotic DNA cleavage that results in double-stranded DNA breakage can be detected well by our neutral comet assay, but that DNA breakage under necrotic conditions involves other types of DNA damage that are less sensitive to detection by comet. The conditions we tested were extreme conditions that induce a typical necrotic type of cell death. It is expected, however, that with milder cell damage some cells might show characteristics of both apoptosis and necrosis, and might therefore appear with longer comet tails similar to those of apoptosis, because these types of cell death form a continuous spectrum of death entity (Portera-Cailliau et al. 1997). It is also possible that these dual features could be seen when apoptotic cells become necrotic at a later stage (Simm et al. 1997).
Although our focus on comet assay for Jurkat cells used the same time window that we had examined by EM, it remains to be determined what chronological profile the results of comet assay would show at later time periods. We plan to research detailed chronological follow-up in a future project. It is also critical to check whether the comet assay yields the same results under different rigors of treatment.
Not many studies have used the comet method for study of apoptosis in parenchymal tissues because of inconsistencies resulting from contamination by tissue debris. We initially tried to perform neutral comet assay on cryosectioned tissues. Although we identified clearly distinct changes in the tissues from burned rats or from ischemia/reperfusion injury, the fluorescence microscopic images were not acceptably analyzable (data not shown). The affected muscle samples showed comet tails, but the images on the microscope were blurry and hazy both because of the high density of nuclei and the presence of too much cytoskeletal and contractile proteins. Hence, we combined comet assay with a method to purify muscle nuclei so that we could apply the comet assay to the study of apoptosis in muscle samples from tissues in vivo.
When purified nuclei from muscle tissues were used, comet assay gave results consistent with those of TUNEL assay, in that the comet score was high for muscle from rats with burns and low for muscle from rats with sham burns. With cardiac muscles from the ischemia/reperfusion model, the comet score was high with the ischemia/reperfused cardiac muscle and low with control and intact parts of the cardiac muscles. These tissues also showed the same patterns with TUNEL staining. Our previous studies in the burn model had used the DNA ladder assay (Yasuhara et al. 2001). The DNA ladder, TUNEL, and comet assay consistently detect apoptosis. From the previous studies, we found that apoptosis starts from 12 hr after burn injury and peaks at day 1 and day 3. In this study, consistent with what was expected, we demonstrated that the purified nuclei from these tissues show enhanced comet tailing, typical of apoptosis. The results of this study imply that the comet assay can be utilized for the detection of apoptosis in skeletal and cardiac muscle tissues.
Although our Jurkat cell models show typical patterns of either apoptosis or necrosis, there are also a number of other stimulations that cause cell death characteristic of both apoptosis and necrosis. In fact, accumulating data from recent reports suggest that apoptosis and necrosis are two extremes on the continuous spectrum of cell death patterns (Portera-Cailliau et al. 1997). There is also cell death with apoptotic cell nuclei with necrotic cytosol, and vice versa. Moreover, at the very late phases of cell death, it is often difficult to distinguish between the two forms, because necrotic cells also give DNA breakage and apoptotic cells will also lead to membrane rupture and organelle damage unless they are phagocytosed. Therefore, our data do not exclude the possibility that necrosis co-exists in the tissues examined. Previous reports indicate that even normal samples can result in a positive comet score, and with a small amount of DNA damage the low comet scores can be reversed to normal (Banath et al. 1998). Therefore, in the study of the quantitation of apoptosis, we counted comet scores of more than 2 for the comparison of comet assay and TUNEL staining. Our results suggest that the comet assay can be an additional tool for selection of assays for the detection of apoptosis in tissues. Although it remains to be established how to correlate the comet scoring with the evaluation of an atypical apoptosis or necrosis, this study has demonstrated that neutral comet assay is a powerful tool to distinguish typical apoptotic and necrotic changes in nuclei, particularly in single cells and possibly in parenchymal tissues.
Footnotes
Acknowledgements
We thank H. Fink for critical advice, B. Crowther for technical assistance with electron microscopy, and R. Khiroya, F. Choles, and C. Mani for their support for the entire project.