
Abstract
The membrane-impermeable chelator CaEDTA was introduced extracellularly among neurons in vivo and in vitro for the purpose of chelating extracellular Zn2+. Unexpectedly, this treatment caused histochemically reactive Zn2+ in intracellular compartments to drop rapidly. The same general result was seen with intravesicular Zn2+, which fell after CaEDTA infusion into the lateral ventricle of the brain, with perikaryal Zn2+ in Purkinje neurons (in vivo) and with cortical neurons (in vitro). These findings suggest either that the volume of zinc ion efflux and reuptake is higher than previously suspected or that EDTA can enter cells and vesicles. Caution is therefore warranted in attempting to manipulate extracellular or intracellular Zn2+ selectively.
R
Materials and Methods
In the first experiment, adult male Sprague-Dawley rats (300–350 g) were anesthetized with halothane, mounted in a stereotaxic device, given a single intraventricular injection of CaEDTA (100 mM, 5 μl, 1 μl/min; P 1.0; L1.5; 2 mm sub dura). After treatment with local anesthetic and suturing the scalp wound, rats were removed from the stereotaxic device and allowed to survive for 10, 20, 60, or 90 min (n=2 per time point), then sacrificed by decapitation under halothane anesthesia. Brains were quickly removed, frozen, cut, stained with TSQ, and imaged, as described previously (Frederickson et al. 1987).
In the second experiment, an NO∗ donor (Spermine-NONOate) was injected into the neocerebellar cortex of the rat (100 mM; 2 μl; 0.4 liter/min) to induce mobilization of Zn2+ from proteins in the perikarya of Purkinje neurons. Each microinfusion solution contained a NO∗ donor (Spermine-NONOate; Sigma, St Louis, MO) that was combined with either saline (NO∗ group; n = 2), saline plus ZnEDTA (control group; n=2), or saline plus the extracellular zinc chelator CaEDTA (5 mM for both EDTA groups; n=2). Animals were allowed to survive for 2 hr after microinfusion, then anesthetized with halothane and decapitated. Brains were removed and immediately frozen.
For quantification of vesicular zinc, frozen brains were cut at 30 μm (- 14C) from the rostral end, and every third section (starting at 3.6 post bregma) was thawed onto a clean glass slide and stained by immersion in a 4.5 μM TSQ solution in 140 mM sodium barbital and 140 mM sodium acetate buffer (pH 10–10.5) for 60 sec (Frederickson et al. 1987). Sections were then rinsed for 60 sec in normal saline and immediately viewed and imaged using a compound fluorescence microscope (Olympus IX 70; excitation 360 nm, emission 500 nm longpass) with a SPOT II cooled CCD 1.4 megapixel camera. All sections obtained from an individual brain (typically five to ten) were used for quantification.
In the third experiment, mixed cortical cell cultures containing both neuronal and astroglial cells were prepared as described previously (Koh et al. 1995) from fetal mice at 14–16 days’ gestation. Dissociated cortical cells were plated onto a previously established astroglial cell monolayer at 2.5 hemispheres per 24-well plate (Nunc) in a plating medium consisting of minimal essential medium (MEM, Earle's salt, glutamine-free) supplemented with 20 mM glucose, 38 mM sodium bicarbonate, 2 mM glutamine, 5% fetal bovine serum, and 5% horse serum. Ten μM cytosine arabinoside was added 5–6 days after cell plating to inhibit the growth of non-neuronal cells. Cultures were maintained at 37C in a humidified CO2 (5%) incubator and used for experiments between 10–13 days in vitro (DIV).
Tissue cultures were exposed to 400 μM zinc for 20 min using MEM at room temperature. After a thorough washout of zinc, cultures were incubated in MEM in the absence or presence of 1 mM CaEDTA or ZnEDTA and placed back in the 5% CO2 incubator.
Intracellular chelatable zinc was visualized in cultured neurons with TFL-Zn, a zinc-specific fluorescent dye (TefLabs; Austin, TX.). Cortical cultures were stained for zinc by adding 50 μM TFL-Zn to the medium, then examined under a fluorescence microscope (excitation 355–375 nm; dichroic 380 nm; barrier 420 nm) (Olympus; Tokyo, Japan), and photographed.
Cell death was morphologically estimated 24 hr after zinc exposure by staining cultures with 0.4% trypan blue to identify dead cells. Lactate dehydrogenase (LDH) release into the bathing medium was used to assess cell injury (Koh and Choi 1987; Koh et al. 1995). Each LDH value, subtracted from the mean background LDH value, was scaled to the mean LDH release (= 100) produced by 24-hr exposure to 300 μM NMDA in sister cultures, where near complete neuronal death with no glial damage occurs. Values greater than 100 indicate glial cell injury in addition to complete neuronal death.
Results
CaEDTA caused an apparent bleaching or depletion of Zn2+ from all cells and all intracellular compartments in all three experiments.
Rats that received CaEDTA in the ventricle and were allowed to survive for various periods thereafter displayed a consistent pattern of reduced staining of vesicular zinc. Specifically, the intensity of staining for vesicular zinc in the hippocampal formation adjacent to the infusion site showed an immediate (within 10 min) decrease (Figure 1), falling further over the next 60 min, then gradually returning to control levels within 90 min. Regions farther from the infusion site followed the same general pattern but with a slightly slower initial decrease in zinc levels (Figure 1).
Similarly, when the cerebellar Purkinje neurons of rats were made zinc-positive by cerebellar infusion of NO∗ (Figure 2B), the CaEDTA co-infusion removed virtually all of the TSQ-positive zinc from those Purkinje somata (Figure 2D). Co-administration of ZnEDTA, in contrast, had no observable effect on the Purkinje somata (Figure 2C).
Finally, in the cultured neuron experiments, cells exposed to high concentrations of zinc could be rescued from zinc toxicity and relieved of their intracellular burden of excess zinc by post-treatment with the extracellular zinc chelator CaEDTA (Figure 3). As shown in Figure 3, treatment with CaEDTA of cells previously loaded with Zn2+ produced a change from TFL-Zn-positive (zinc-loaded) to TFL-Zn-negative (zinc-free) staining. One hour after zinc exposure, most neurons exhibited markedly increased fluorescence in their cell bodies and processes (Figure 3A). However, when cultures were incubated with 1 mM CaEDTA, the intensity of zinc fluorescence was equivalent to the intensity of sham-rinsed controls (Figure 3B). ZnEDTA was unable to chelate any additional zinc, thus having limited effects on the intensity of TFL-Zn staining (Figure 3C). Estimation of cell death 24 hr after zinc exposure revealed that 1 mM CaEDTA, but not ZnEDTA, blocked zinc-induced cell death, verified by trypan blue staining (Figures 3D-3F) and LDH release assay (Figure 3G).
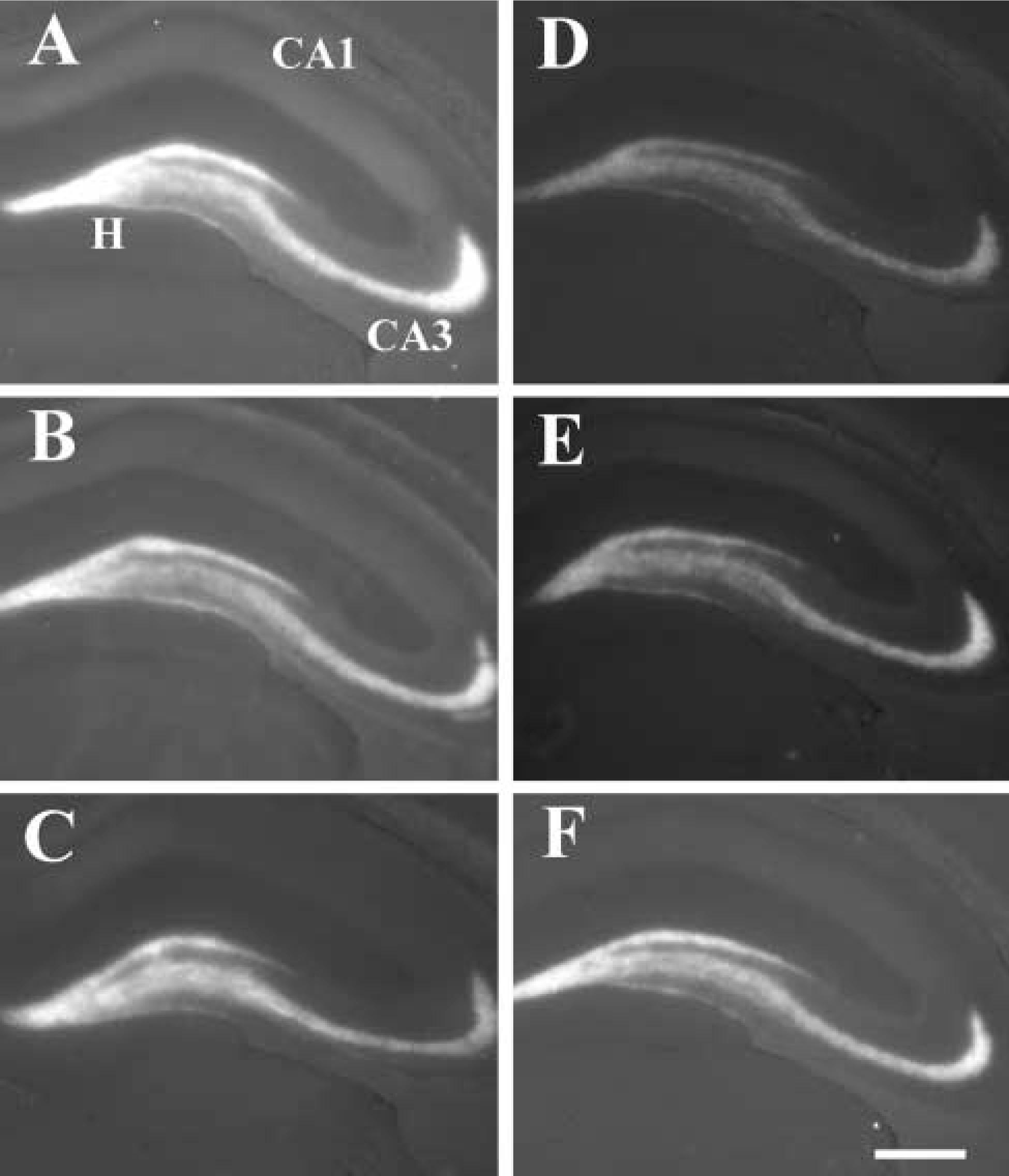
Zinc staining (TSQ fluorescence) of the hippocampus. (
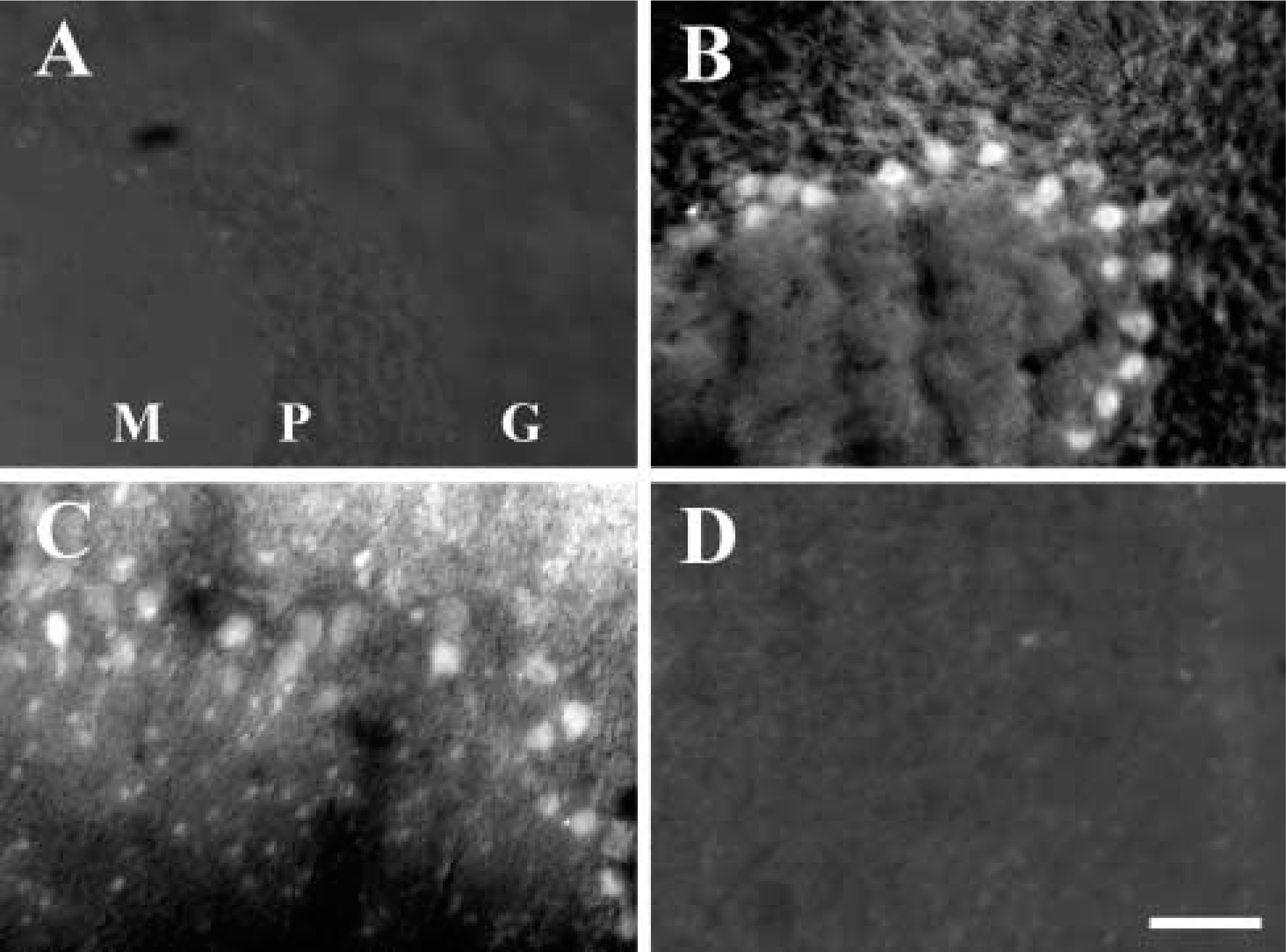
Zinc staining (TSQ) in the cerebellum of sham-operated animals (
Discussion
The central finding of this study is that the nominally membrane-impermeable chelator CaEDTA apparently depletes intracellular pools of stainable or free Zn2+. In cases in which neurons were loaded with stainable zinc in the perikaryon, i.e., the Purkinje cells in vivo and the dissociated cortical neurons in vitro, the intracellular Zn2+ burden represented a pathological Zn2+ load, induced either by Zn2+ “poisoning,” (Frederickson 1989), adding excess zinc to the medium of the cultured neurons (Yokohama et al. 1986), or by probable mobilization of free zinc from intracellular proteinaceous stores (Cuajungco and Lees 1998) in the case of the NO∗-exposed Purkinje cells. Despite the different origins of free Zn2+, the presence of the CaEDTA in the extracellular fluid reduced the Zn2+ burden.
Presumably, the zinc ions inside cells can efflux through the same gated channels that allow influx, i.e., the voltage- and glutamate-gated channels described by Weiss and co-workers (Sensi et al. 1997; Weiss et al. 2000). By preventing re-entry and by maintaining a steep Zn2+ gradient across the membrane, CaEDTA appears to “pull” the free Zn2+ from exposed neurons. The Kd for Zn2+ binding to EDTA is 10−15.5 M (Cuajungco and Lees 1997) thus, with a molar excess of the EDTA. Free Zn2+ in the extracellular medium would be about 1 pM in the presence of EDTA. Zinc ions leaving neurons down their gradient would presumably be trapped by the CaEDTA, thus slowly depleting the neuronal content.
In the case of the vesicular Zn2+, the effect of CaEDTA might be similar to the effects seen within the zinc-loaded somata. Therefore, CaEDTA in the extracellular fluid would capture any Zn2+ released from the presynaptic boutons, either by regulated or by constitutive exocytosis or by other release mechanisms. Hence, trapped by CaEDTA in the cleft, Zn2+ would be unavailable for reuptake into the boutons and would gradually deplete the axon boutons of their pools of vesicular zinc.
It is also possible that CaEDTA interferes directly with the TSQ staining procedure. For example, tissue saturated with CaEDTA is cut in the cryostat and stained immediately with TSQ, thereby allowing the two zinc chelators to compete for the ions. Although this artifactual suppression of the TSQ staining cannot be completely ruled out, it cannot account for the rescue of the cultured neurons. More specifically, in experiment 3 the neuronal release of LDH was prevented by CaEDTA, an effect that cannot be attributed to chelator-chelator interactions. Similarly, the fact that CaEDTA suppression of vesicular zinc staining was essentially complete by the time (2 hr after injection) that the suppression of the NO∗-induced staining was complete, argues against postmortem chelator-chelator interactions as an explanation for the present findings.
Taken together, the present experiments provide evidence of more rapid and abundant transit of Zn2+ across neuronal membranes than was previously believed. These effects might be explained by the activity at effluxing pumps, such as ZnT-1 (Palmiter and Findley 1995), or the reversible plasma membrane transporter found in membrane vesicles (Colvin 1998), or via the zinc-permeable channels that have been characterized (Weiss et al. 2000). One interesting question is whether the efflux of Zn2+ from somata depends on some of the same zinc-permeable channels involved in Zn2+ influx. If so, the depletion of intracellular zinc by an extracellular chelator could perhaps be modulated by the same ligands (e.g., KA, AMPA, NMDA) that modulate Zn2+ influx (Weiss et al. 2000).
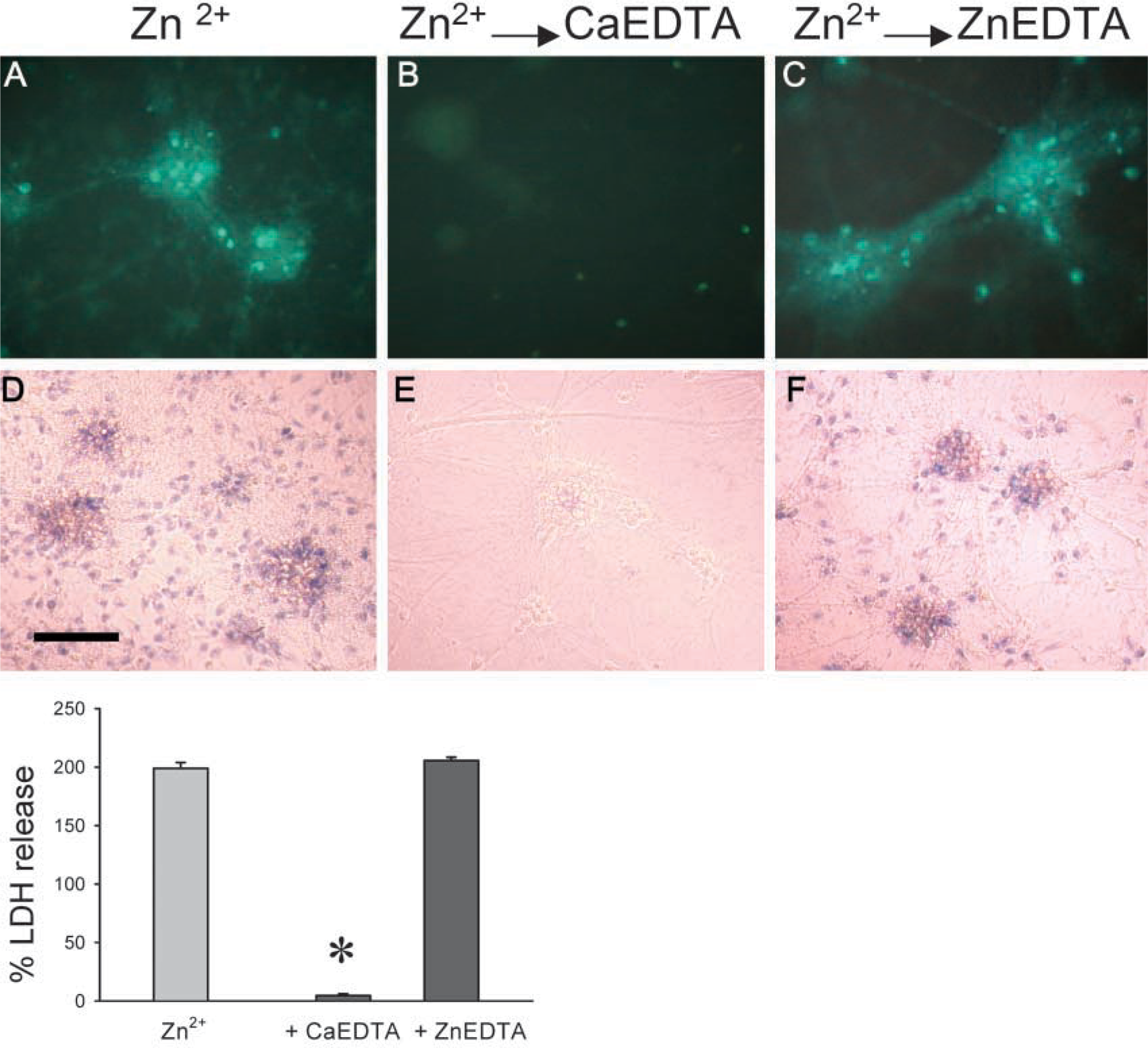
Zinc staining in cortical cultures incubated for 1 hr in MEM (
Footnotes
Acknowledgements
Supported in part by NS40215, NS42882, and 46,682 to CJF, N538535 to RBT, and by Creative Research Initiatives of Korea Ministry of Science and Technology to JY.
The authors gratefully acknowledge the skillful technical assistance of Yaping Zeng and Cathy Frederickson.