
Abstract
The putative anion channel mCLCA3 (alias gob-5) is the third murine member of the recently discovered family of calcium-activated chloride channels (CLCA family). Preliminary data suggest that mCLCA3 may play a significant role in diseases with secretory dysfunctions, including asthma and cystic fibrosis. In this study, the mCLCA3 protein was characterized biochemically and its cellular and subcellular distribution pattern was established in normal murine tissues. Polyclonal rabbit antibodies were generated and affinity-immunopurified using synthetic oligopeptides corresponding to the extracellular amino terminus of the mCLCA3 polypeptide. After in vitro translation and glycosylation, protein-ase K protection assay, and heterologous expression in COS-7 or HEK 293 cells, SDS-PAGE and immunoblotting revealed a protein structure similar to that of previously characterized CLCA proteins. A systematic light, confocal laser scanning, and transmission electron microscopic immunolocalization study, including virtually all murine tissues, identified the mCLCA3 protein exclusively associated with mucin granule membranes of gastrointestinal, respiratory, and uterine goblet cells and other mucin-producing cells. The results suggest that mCLCA3 may be involved in the synthesis, condensation, or secretion of mucins.
A
The third murine CLCA homologue, mCLCA3 (alias gob-5), has been identified in goblet cells throughout the intestinal tract using in situ hybridization (Komiya et al. 1999). The message was also detected by Northern blotting hybridization in the murine trachea and uterus without identification of the respective cell types. Functional analyses of mCLCA3 have not been reported to date and the biological processes in which it is involved are unclear.
Increasing evidence suggests that members of the CLCA family play a role in diseases with epithelial secretory dysfunctions. For example, in a murine model of bronchial asthma, mCLCA3 was found to be a key regulator in the induction of mucus overproduction. In this model, antisense mCLCA3 therapy effectively suppressed the asthma phenotype, whereas mCLCA3 overexpression exacerbated the condition (Nakanishi et al. 2001). In a related in vitro model, introduction of mCLCA3 into the mucoepidermoid cell line NCI-H292 induced mucus production and expression of MUC family members. Upregulation of mCLCA3 was found in transgenic mice overexpressing IL-9, which develop a similar asthma-like phenotype (Zhou et al. 2001). In these mice, neutralizing IL-9 antibody treatment suppressed the expression of mCLCA3 mRNA, whereas IL-9 treatment of wild-type mice enhanced its expression. Excess mucus production is also a hallmark of cystic fibrosis (CF) in several murine Cftr (cystic fibrosis transmembrane conductance regulator) knockout models. CF mice usually die before or around weaning due to ileal obstruction resulting from intestinal goblet cell hyperplasia and excessive production of a stringy mucus (reviewed by Grubb and Boucher 1999). An as yet unidentified calcium-activated chloride conductance is believed to play a major role as a modulator of the CF phenotype by partially compensating for the basic chloride secretory defect in the absence of the CFTR chloride channel (Clarke et al. 1994; Rozmahel et al. 1996). The functional characteristics of CLCA proteins (reviewed by Fuller and Benos 2000) and their expression in the digestive and respiratory tracts raise the question of how CLCA family members are involved in the pathophysiology of these conditions. In particular, mCLCA3 and its human orthologue hCLCA1 are promising candidates because of their expression in intestinal goblet cells (Gruber et al. 1998a; Komiya et al. 1999).
Knowledge of the cellular and intracellular locations of a channel protein is essential for understanding the biological and pathophysiological processes in which it is involved. On the subcellular level, it is of particular interest whether a channel is expressed in the apical or basolateral cell membrane, or in the membranes of specific organelles. Moreover, certain members of the CLCA gene family (Lu-ECAM-1 clone 4, hCLCA3) appear to be totally secreted proteins (Elble et al. 1997; Gruber and Pauli 1999), a scenario that was also conceivable for the goblet cell-specific mCLCA3. To resolve this question, comprehensive immunolocalization studies were performed in this study on the light microscopic and ultrastructural levels. The mCLCA3 protein was detected in the membranes of mucin granules of intestinal, respiratory, and uterine goblet cells and other mucin-producing cells, suggesting that it takes part in the synthesis, condensation, or secretion of mucus.
Materials and Methods
Animals
Adult male and female C57BL/6 and NMRI mice were obtained from the Institute for Laboratory Animal Science and Central Animal Facility, Medical School Hannover, and were sacrificed in accordance with national guidelines.
Cloning of the mCLCA3 cDNA
The mCLCA3 open reading frame (ORF) was cloned and sequenced from murine large intestinal mRNA using an RT-PCR-based strategy. In brief, total RNA was isolated from colon tissue using the Trizol method (Invitrogen; Carlsbad, CA) and selected for poly-A tailed mRNA using the Nucleotrap mRNA Kit (Macherey-Nagel; Düren, Germany). The mRNA was reverse-transcribed using poly-T primers and Superscript RNase H reverse transcriptase (Invitrogen), followed by incubation with RNase H (Sigma; St Louis, MO). Based on the cDNA nucleotide sequence accessible in the GenBank database (accession no. AB016592), oligonucleotide primers flanking the mCLCA3 ORF were designed for primary PCR (upstream primer 5′-GAAAGCTGCAGGATGGAATC-3′; downstream primer 5′-GACTGGTTGATTTCTTGCCTG-3′) and for nested PCR containing linkers with NotI restriction sites (upstream primer 5′-ATGCGGCCGCGATGGAATCTTTGAAGAGTCCTG-3′; downstream primer 5′-ATGCGGCCGCTCAGTGCAAACCT-AGTGTCACC-3′, with NotI sites in italics). Both PCR steps were performed using Pwo DNA polymerase (Roche Molecular Biochemicals; Indianapolis, IN) and the following conditions: 25 (primary PCR) or 35 (nested PCR) cycles of 95C for 1 min, 56C (primary PCR) or 58C (nested PCR) for 40 sec, and 72C for 90 sec with a time increment of 8 sec per cycle for each extension step (72C), followed by a final extension at 72C for 10 min. PCR products were gel-purified, incubated with NotI, and cloned into the NotI site of pcDNA3.1 (Invitrogen). The vector insert was sequenced and compared with the published cDNA sequence to verify identity of the clone and to exclude sequence errors introduced by PCR.
In Vitro Translation and Protease Protection Assay
The mCLCA3 ORF in pcDNA3.1 was transcribed and translated with the TNT T7 Coupled Reticulocyte Lysate System (Promega; Madison, WI). Reactions of 25 μl were carried out at 30C for 90 min without or with (2 μl) canine pancreatic microsomal membranes (Promega). Samples were analyzed by 10% SDS-PAGE and immunoblotting as described below. In addition, protease protection assays were performed as described (Gruber et al. 1999). Briefly, in the presence of microsomal membranes, in vitro-translated mCLCA3 was incubated with 0.4 μg/μl proteinase K (Sigma) for 60 min on ice. The reaction was stopped by boiling and was analyzed using 13% SDS-PAGE and immunoblotting as described below.
Heterologous Expression in Mammalian Cells
The mCLCA3 ORF cloned in pcDNA3.1 was transfected into 70% confluent human embryonic kidney cells (293 cells) or COS-7 cells using the Polyfect method (Qiagen; Hilden, Germany) according to the manufacturer's protocol. The cells were harvested 48 hr after transfection and lysed as described below. As controls, cells were transfected using the same protocol and pcDNA3.1 containing the hCLCA2 ORF or with the transfection reagent alone (mock transfection).
Generation of Antibodies Against mCLCA3
Regions of high predicted immunogenicity were selected from the mCLCA3 polypeptide using computer-aided antigenicity analysis. A GenBank search was performed to exclude homologies of the selected regions with other known proteins and in particular with the related murine CLCA homologues mCLCA1 and mCLCA2. Two oligopeptides were synthesized (p3a, corresponding to amino acids 83 to 97, ESWKAKPEYTRPKLE, and p3b, corresponding to amino acids 253 to 267, EKNHNQEAPNDQNQR), conjugated to keyhole limpet hemocyanin (KLH), and used for standard immunization of two rabbits each. Pre-immune sera were collected before immunization and used as controls in the immunodetection experiments. The four antisera were designated α-p3a1, α-p3a2, α-p3b1, and α-p3b2. Immune sera α-p3a1 and α-p3a2 were affinity-immunopurified using the peptide p3a coupled to an EAH-Sepharose column.
Native Tissue Sample Preparation and Immunoblotting
Fresh tissue samples from murine kidney, small intestinal mucosa, and large intestinal mucosa were lysed in 50 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Triton X-100, and 0.5% sodium desoxycholate in the presence of protease inhibitors (1 mM phenylmethanesulfonyl fluoride, 1 μg/ml pepstatin, 5 μg/ml leupeptin, 5 μg/ml antipain, and 1 μg/ml aprotinin). Transiently transfected 293- and COS-7 cells were lysed accordingly. SDS-PAGE (10% or 13%) was performed in the presence of 2 mM DTT according to standard protocols. After electroblotting onto nitrocellulose membranes and blocking of the membranes with Tris-buffered saline (TBS) containing 0.1% Tween-20 and 5% non-fat milk, membranes were probed at 4C overnight with the purified antibodies α-p3a1 (1.36 mg/ml stock) or α-p3a2 (0.74 mg/ml stock) or with the immune sera α-p3b1 or α-p3b2 diluted in blocking buffer (dilutions ranging from 1:100 to 1:4000). Membranes were then incubated with horseradish peroxidase-conjugated swine anti-rabbit immunoglobulins (0.4 μg/ml; Dako, Carpinteria, CA) and developed using enhanced chemiluminescence (Amersham; Arlington Heights, IL). For negative controls, membranes were probed with the respective pre-immune serum instead of immune serum or purified antibodies.
Immunohistochemistry
Fresh murine tissue samples were fixed in 4% neutral buffered formaldehyde and routinely embedded in paraffin or fixed in 4% neutral buffered formaldehyde containing 0.1 M cacodylic acid and routinely embedded in glycolmethacrylate (GMA). The following organs and tissues were processed: liver, gallbladder, spleen, kidneys, urinary bladder, pancreas, oral cavity, parotid and sublingual salivary glands, esophagus, stomach, duodenum, jejunum, ileum, cecum, colon, rectum, nasal cavity, trachea, lung, heart, adrenal glands, thyroid glands, ovaries, oviducts, uterus, cervix, vagina, mammary glands, testes, epididymides, male accessory sex glands (seminal vesicular glands, coagulating glands, ampullary glands, dorsal and ventral prostates, bulbourethral glands, preputial glands), lymph nodes, brain (cortex, cerebellum, brainstem, medulla), eyes, skin, adipose tissue, skeletal muscle, bone, and large vessels (thoracic and abdominal aorta, vena cava). Paraffin-embedded tissues were cut at 3 μm and GMA-embedded tissues were cut at 1 μm. Tissue sections were mounted on SuperfrostPlus adhesive glass slides (Menzel-Gläser; Braunschweig, Germany). In addition to the immunohistochemical (IHC) analyses, consecutive tissue sections were routinely stained with hematoxylin and eosin (HE) for histological examination and with periodic acid-Schiff (PAS) reaction or alcian blue to stain the mucins. For IHC staining of paraffin-embedded tissue sections, the avidin-biotin-peroxidase complex (ABC) method was applied. After dewaxing of the mounted tissue sections in xylene and rehydration in isopropanole and graded ethanol, the following antigen retrieval (AR) methods were tested: (a) 15-min microwave heating (700 W) in 10 mM citric acid, pH 6.0, (b) 20-min treatment with 0.05% pronase E (Merck; Darmstadt, Germany) in PBS at 37C, and (c) microwaving after pronase E digestion. Due to superior results of the microwave-only treatment, method a was used for the systematic tissue analyses. Endogenous peroxidase activity was inhibited by incubating the slides with 70% methanol containing 0.5% H2O2, followed by washes in PBS containing 0.05% Tween-20 (PBS/Tween-20) and blocking in PBS/Tween-20 containing 20% heat-inactivated normal goat serum. After repeated washes, the sections were incubated with the purified antibodies α-p3a1 or α-p3a2, the specific immune sera α-p3b1 or α-p3b2, or the respective pre-immune sera in PBS/Tween-20 containing 1% BSA (dilutions ranging from 1:100 to 1:32,000) in a humid chamber at 4C overnight. Sections were washed in PBS/Tween-20 and incubated at room temperature (RT) for 30 min with biotinylated goat anti-rabbit immunoglobulins (5 μg/ ml; Vector Laboratories, Burlingame, CA) diluted in PBS/ Tween-20, followed by repeated washes in PBS/Tween. Color was developed for 30 min using freshly prepared ABC solution (Vectastain Elite ABC Kit; Vector Laboratories) diluted in PBS, followed by repeated washes in PBS and rinsing in tapwater. The slides were counterstained with hematoxylin, dehydrated through ascending graded ethanol, cleared in xylene, and coverslipped. GMA-embedded tissue sections were processed using the indirect peroxidase method. IHC staining was performed as described above with two minor modifications: dehydration was performed in methanol and peroxidase-conjugated swine anti-rabbit immunoglobulins (6.5 μg/ml; Dako) were applied as secondary antibodies. For confocal laser scanning microscopy, both paraffin- and GMA-embedded tissue sections were processed as described above, with minor modifications. Rhodamine-conjugated goat anti-rabbit immunoglobulins (5 μg/ml; Dianova, Hamburg, Germany) were applied at RT for 30 min as secondary antibodies. Sections were coverslipped using an aqueous mounting medium without previous counterstaining. The sections were examined with a Leica TCS SP2 confocal laser scanning microscope (Leica; Solms, Germany) equipped with helium and neon lasers. Rhodamine was excited at 530 nm and emission of red fluorescence was detected at 590 nm. Single optical scans were repeated several times and recorded separately to be subsequently combined as an average of three images.
Immune Transmission Electron Microscopy
Fresh tissue specimens of approximately 4 mm3 were fixed at 4C overnight in 4% neutral buffered paraformaldehyde containing 0.25% glutaraldehyde and 3.7% saccharose. After five 20-min washes at RT in 0.1 M cacodylic acid, samples were dehydrated at −20C through ascending graded ethanol in six 30-min steps and gradually infiltrated at −20C with LR White resin (medium grade; London Resin, Berkshire, UK; 100% ethanol: LR White 2:1, 1:1, and 1:2 for 30 min each, 100% LR White for 30 min and overnight). The specimens were embedded in gelatin capsules filled with LR White resin containing LR White accelerator (Polysciences; Warrington, PA), and resin was allowed to polymerize at −20C overnight under exposure to UV light. Ultrathin sections cut at 60–90 nm were collected on uncoated 200-mesh nickel grids. For immunolabeling, grids were immersed upside down in drops of the respective solution. Aldehydes were blocked in four 5-min steps with PBS containing 50 mM glycine. The sections were blocked for 15 min in PBS containing 0.5% BSA and 0.1% liquid gelatin (washing buffer) and 5% heat-inactivated normal goat serum. Incubation with the purified antibodies α-p3a1 or α-p3a2, the immune sera α-p3b1 or α-p3b2, or the respective pre-immune sera was performed in washing buffer at 4C overnight in a humid chamber (dilutions ranging from 1:100 to 1:8000), followed by 15 2-min washes in washing buffer and by incubation for 1 hr with 10-nm gold particle-conjugated goat anti-rabbit immunoglobulins (8.38 × 1012 gold particles/ml; Sigma) diluted at 1:40 in washing buffer. After repeated washes in washing buffer and subsequently in PBS, sections were postfixed for 5 min in PBS containing 2% glutaraldehyde. The grids were rinsed in distilled water, followed by contrasting for 15 min in aqueous uranyl acetate (Ultrostain 1; Leica), and final washes in distilled water. The grids were air-dried and tissue sections were examined and photographed with a Zeiss EM 10 A transmission electron microscope (Zeiss; Oberkochen, Germany).
Results
Generation of Antibodies
To obtain specific antibodies for immunolocalization of the mCLCA3 protein in murine tissues, two rabbit immune sera were raised and affinity-immunopurified using a synthetic oligopeptide corresponding to the extracellular amino terminus of the mCLCA3 protein (purified antibodies α-p3a1 and α-p3a2). As control for the specific detection of the mCLCA3 protein, two additional immune sera were raised using a second oligopeptide, corresponding to an adjacent region of the mCLCA3 polypeptide (immune sera α-p3b1 and α-p3b2). In all subsequent immunoblotting, immunohistochemical, and immune transmission electron microscopic analyses, virtually the same specific staining patterns were obtained with the purified antibodies and the immune sera, indicating that in fact the mCLCA3 protein was detected.
Immunoblotting Analyses
Probing of the in vitro-translated mCLCA3 protein using the purified antibodies or the immune sera identified a primary translation product of approximately 100 kD (Figure 1A) that increased in size to approximately 110 kD after glycosylation by microsomal membranes (Figure 1B, left). Addition of proteinase K to the microsomal membranes reduced the size of the protein detected by the antibodies to approximately 35 kD (Figure 1B, right), corresponding to the calculated size of the extracellular amino terminus of the protein. When lysates of transiently mCLCA3-transfected 293 and COS-7 cells were immunoblotted with the purified antibodies or immune sera, a single protein species of approximately 90 kD was detected that was not present in the hCLCA2-transfected or mock-transfected cells (Figure 2). The glycosylated primary translation product of approximately 110 kD was apparent only in transfected COS-7 cells probed with the immune sera (Figure 2B). Probing of murine renal, small, and large intestinal tissue lysates yielded specific bands only in the small and large intestines (Figures 3A and 3B). A protein species of approximately 90 kD was specifically detected in both the small and the large intestine, but it was significantly more abundant in the large intestinal lysate. In addition, a second protein of approximately 45 kD was specifically labeled in the large intestine that was absent in the small intestine. Immunoblotting analyses using the non-purified immune sera were generally complicated by several unspecific bands that were negligible or absent when the immunopurified antibodies were used (compare Figures 2A and 2B and Figures 3A and 3B).
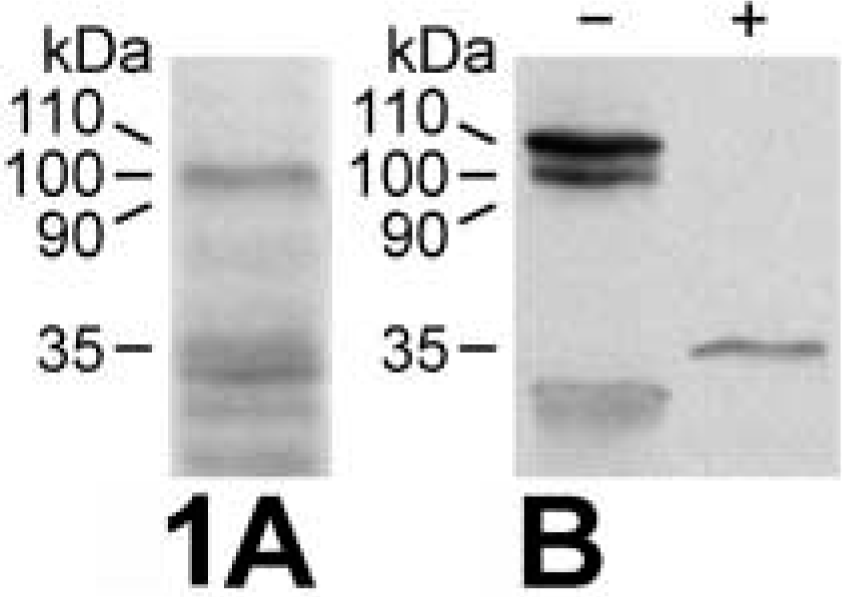
Immunoblotting analyses of the in vitro-translated mCLCA3 protein. (
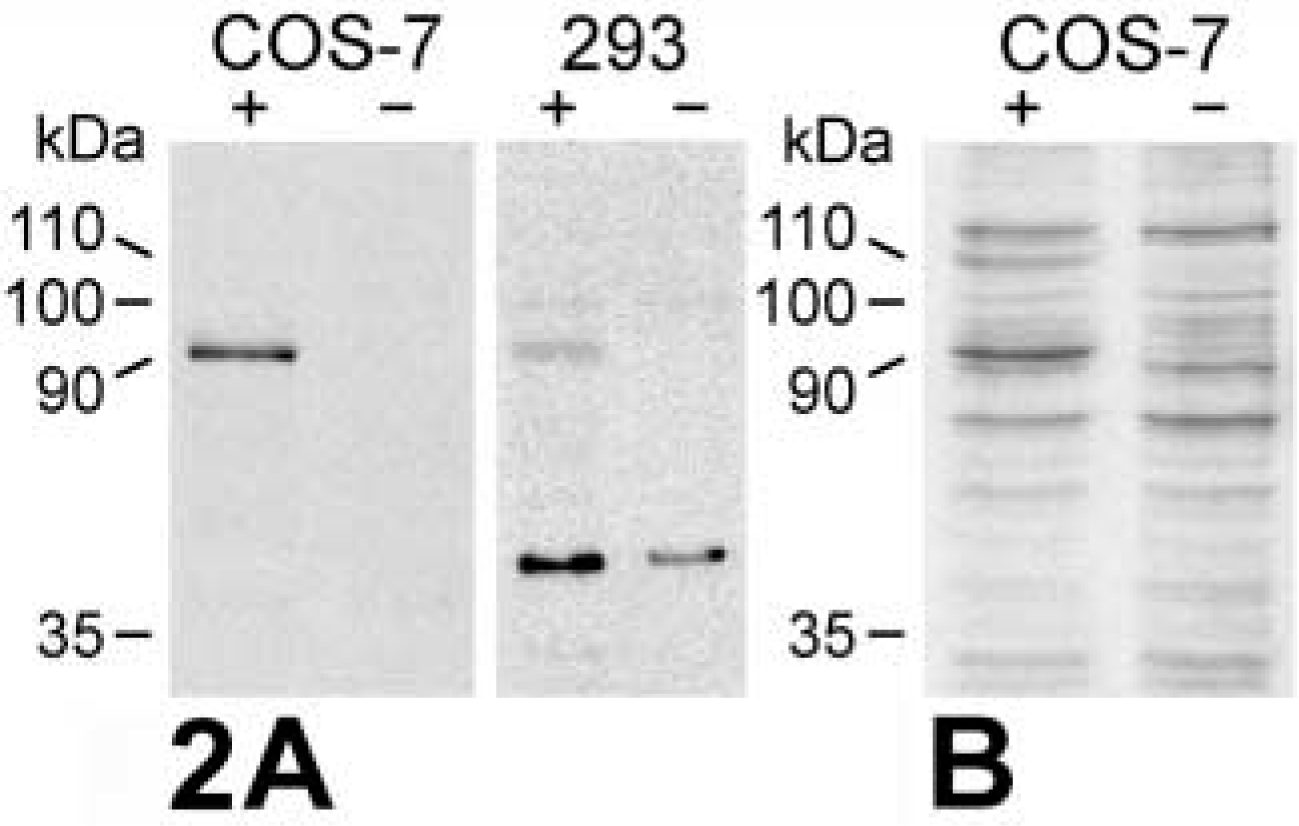
Immunoblotting analyses of mCLCA3-transfected (+) and mock-transfected (-) COS-7 and 293 cells. (
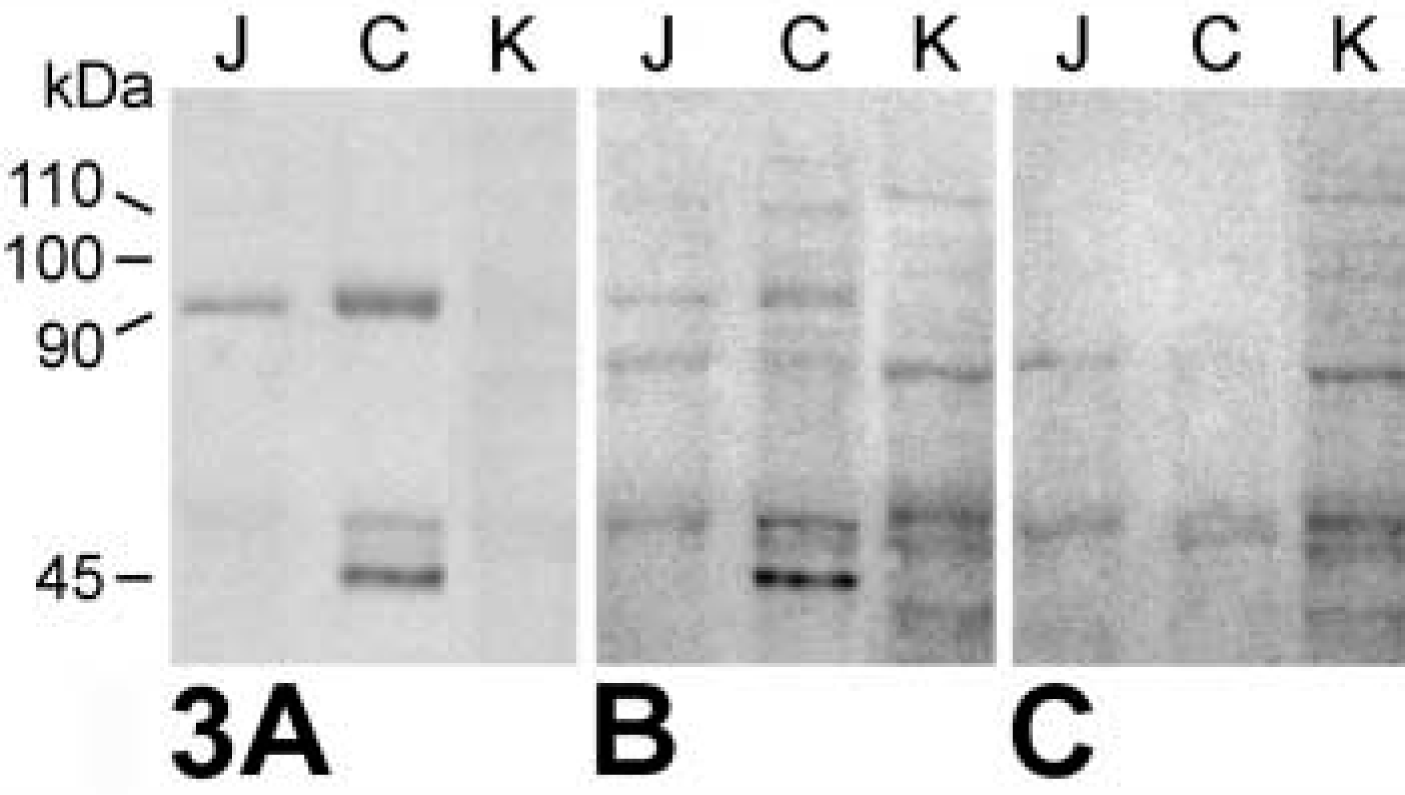
Immunoblotting analyses of lysates from murine jejunal mucosa (J), colon mucosa (C), and kidney (K) using the purified antibodies α-p3a1 (
Tissue and Cellular Distribution Pattern
Of all tissues analyzed by IHC, the mCLCA3 protein was exclusively detected in the digestive and respiratory tracts and in the uterus (Figure 4). No differences were noted among individual animals, and similar staining patterns were obtained when the different antibodies were used. To identify cells that produce mucins, HE, PAS, and alcian blue stains were prepared using consecutive tissue sections (e.g., see Figure 4D). A comparison of these stains with the immunohistochemical stains revealed that mCLCA3 was expressed only in mucin-producing cells. However, not all of the different cell types that secrete mucins were labeled. In the respiratory tract, the epithelia of the trachea and major bronchi contained single or clustered goblet cells that were all intensely labeled (Figures 4A and 4C). Somewhat weaker staining was detected in the submucosal glands of the upper segment of the trachea (Figure 4B). All ciliated epithelial cells were negative. In the uterus, virtually all goblet cells interspersed in the epithelium, as well as mucinous cells of the uterine glands, were positive (Figures 4E and 4F) as confirmed by PAS and alcian blue stains on consecutive tissue sections (not shown). Again, all non-mucinous cells were negative. In the stomach, all surface mucous cells were intensely stained, whereas parietal and chief cells were negative (Figure 4G). All goblet cells were stained in the villi throughout the small intestine, including duodenum, jejunum, and ileum (Figures 4H and 4I). However, crypt goblet cells in the small and large intestine showed staining only in approximately the upper two thirds of the mucosal crypts, whereas weak or no staining was observed in the basal third (Figures 4H-4K). In addition, in both the small and large intestine there was occasional staining of the mucous layer lining the intestinal lumen (Figures 4I and 4J). Enterocytes were negative. Interestingly, other secretory tissues containing mucinous cells, including gallbladder, kidney, pancreas, sublingual salivary glands, oviduct, mammary gland, and prostate, did not express mCLCA3. No labeling was present in any of the tissues when pre-immune sera were used at dilutions that yielded specific staining with the purified antibodies or immune sera (Figure 4L).
Intracellular Localization of the mCLCA3 Protein
In the paraffin-embedded tissue sections, all labeled cells of the different organs exhibited a granular staining pattern throughout the cytosol, with no assignment to a specific cellular structure possible (Figure 5A). A few goblet cells, particularly those in the intestine, had an intensely labeled cap at the luminal surface (Figure 4I). A more detailed intracellular staining pattern was observed using GMA-embedded sections that allow access of the antibodies only to the surface of the tissue that is otherwise impermeable to antibodies. In these sections, labeling was apparent around but not within mucin granules and in a finely granular pattern throughout the cytosol (Figure 5B). Similar to the paraffin-embedded tissues, few cells had intensely stained caps at the luminal surface. Using pre-immune sera as primary antibodies yielded no labeling at dilutions used for the immune staining (Figure 5C). A virtually identical staining pattern was found in GMA-embedded tissue sections using confocal laser scanning microscopy with rhodamine-conjugated secondary antibodies (Figure 5D). Immune transmission electron microscopy using gold-labeled secondary antibodies was performed to localize the mCLCA3 protein on the ultrastructural level. The 10-nm gold particles were intimately associated with the membranes and with peripheral electron-dense zones of small, medium-sized, and large mucin granules throughout the goblet cells (Figures 6A-6C). In addition, electron-dense material budding from the luminal surface of large, mature mucin granules was labeled, usually close to the luminal surface of goblet cells (Figure 6D). Gold particles were not observed in the electron-lucent center of mucin granules, in the cytosol, nucleus, other organelles, or along the basolateral membrane of goblet cells. Occasionally, gold particles were located in electron-dense regions of the mucous layer lining the luminal surface of the intestine (Fig 6E). The intestinal brush border and the electron-lucent, serous parts of the mucous layer were not labeled. Virtually identical labeling patterns were observed using the purified antibodies or the immune sera, and no cellular structures were labeled when the pre-immune sera were used as primary antibodies at the same dilutions.
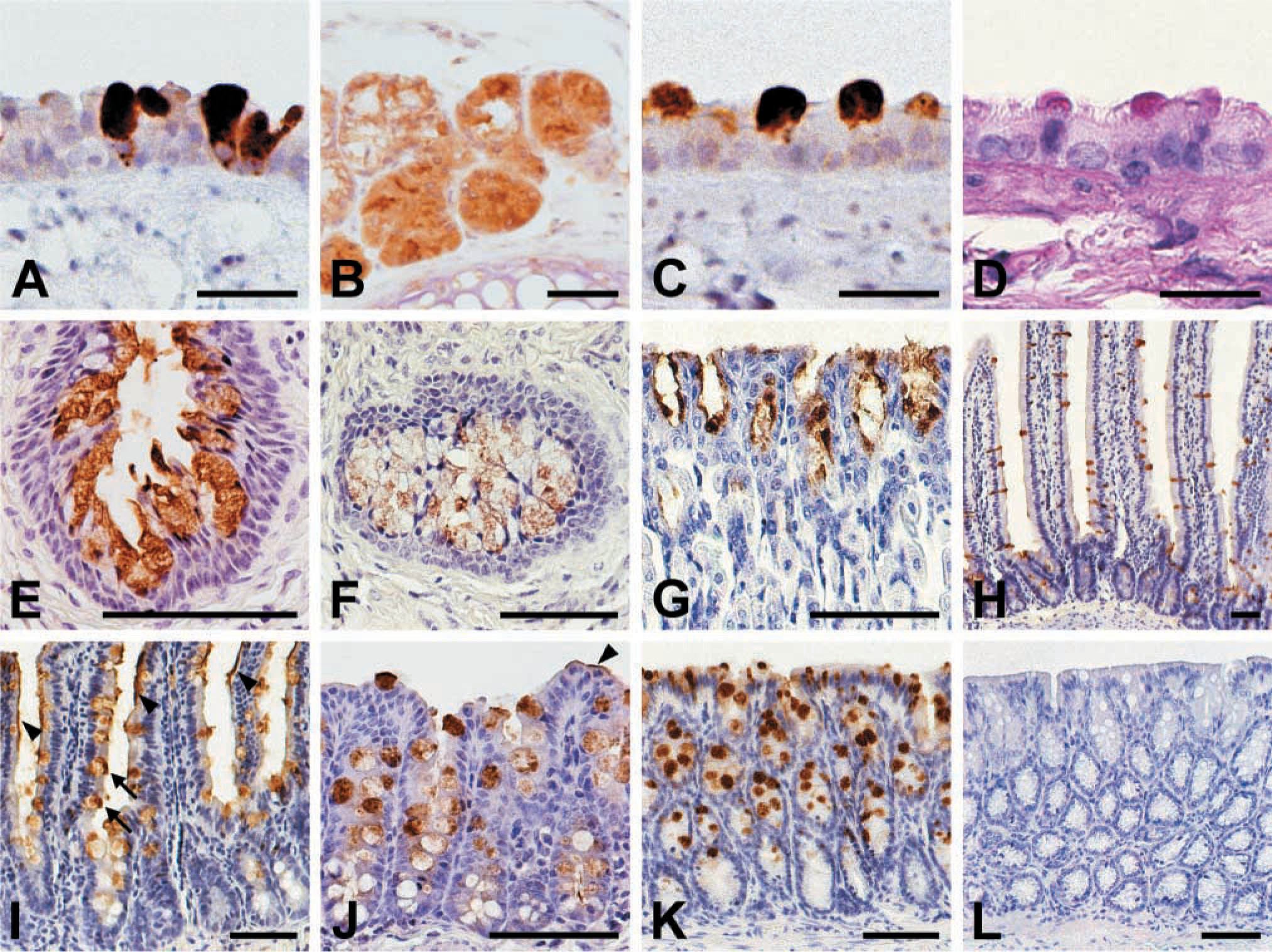
Tissue and cellular distribution pattern of the mCLCA3 protein as detected by immunohistochemistry (brown staining) using formalin-fixed, paraffin-embedded tissue sections from murine trachea (
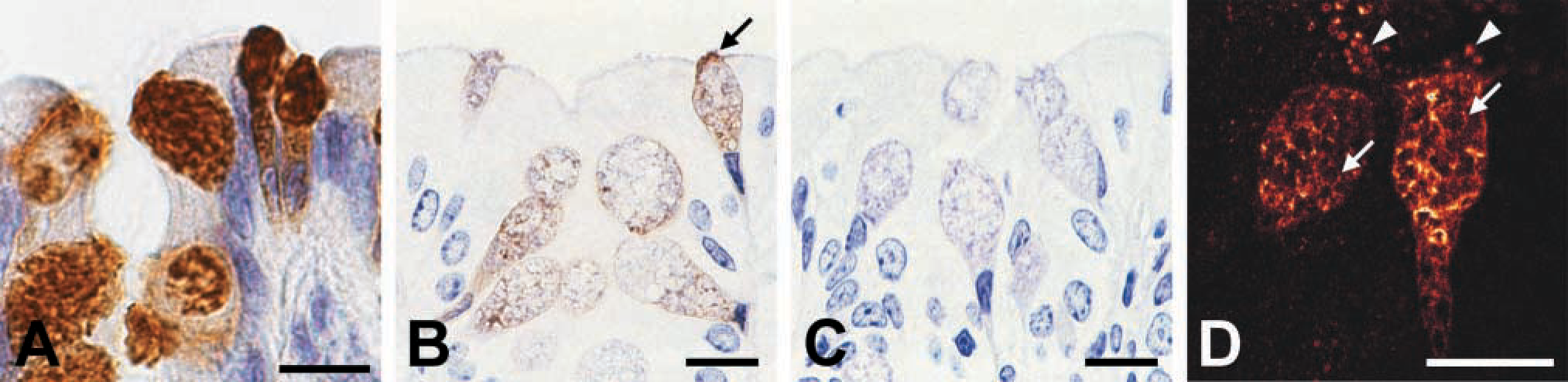
Intracellular distribution pattern of the mCLCA3 protein in intestinal goblet cells as detected by different light microscopic techniques. (
Discussion
The results of the biochemical experiments yield first clues to the structure and processing of the mCLCA3 protein that appear to be similar to those of other CLCA proteins. Accordingly, the glycosylated primary mCLCA3 translation product of approximately 110 kD is processed to an approximately 90-kD amino terminal cleavage product in both transfected mammalian cells and native murine tissues. Moreover, protection of an approximately 35-kD protein fragment from protease digestion after in vitro translation in the presence of microsomal membranes established that mCLCA3 is an integral membrane protein with an intraluminal or extracellular amino terminus. The size of the protected fragment recognized by the antibodies is similar to the size of the extracellular amino termini of all other CLCA homologues analyzed thus far (Elble et al. 1997; Gruber et al. 1998a; Gruber and Pauli 1999). Therefore, the biochemical results obtained in this study are consistent with the proposed general model of CLCA protein structure and processing (reviewed by Gruber et al. 2000). The nature of the additional protein species of approximately 45 kD identified in the large intestinal tissue lysate remains unexplained at this point. Attempts to isolate sufficient pure protein for sequencing have been unsuccessful thus far (data not shown). It appears that this protein fragment represents a truncated portion of the amino terminus of the protein with obvious similarities to the secreted 37-kD amino terminus of hCLCA3 (Gruber and Pauli 1999) and the putatively secreted 32-kD amino terminal clone 4 of Lu-ECAM-1 (Elble et al. 1997). However, other scenarios, including alternatively spliced variants, cannot be excluded, and the function and significance of the 45-kD species of mCLCA3 in the colon remain to be established.
The mCLCA3 protein is exclusively associated with the mucin granule membranes of goblet cells, suggesting that it plays a role in the synthesis, condensation, or secretion of mucins. Several observations indicate that the immunolocalization studies in fact detected the mCLCA3 protein. First, the obtained tissue expression pattern in the digestive and respiratory tracts and in the uterus is consistent with the mCLCA3 mRNA distribution as detected by Komiya and co-workers (1999). Second, antibodies raised against two different segments of the mCLCA3 polypeptide yielded virtually identical staining patterns at both the light microscopic and the ultrastructural level. Third, the localization of the protein in the membranes of mucin granules is consistent with its proposed role in the regulation of mucin secretion (Nakanishi et al. 2001). Fourth, tissues known to express the related murine homologues mCLCA1 (including kidney and testes; Gruber et al. 1998b) and mCLCA2 (mammary gland; Lee et al. 1999) did not stain with the antibodies directed against mCLCA3, thus excluding crossre-activity with closely related CLCA members.
The location of the mCLCA3 protein in goblet cells would be consistent with a contribution to the flux of chloride ions across epithelia containing this cell type. In addition, the presence of a putative chloride channel protein in the membranes of goblet cell mucin granules appears plausible because considerable amounts of water are withdrawn from these vacuoles during their maturation (reviewed by Colony 1996). Furthermore, the negatively charged mucous glycoproteins require strong acidification within the granule compartment to be densely packed. Low pH values are achieved by integral membrane proton pumps (H+-ATPases) and parallel chloride conductance which allows counterion transport, helping to collapse the membrane potential and facilitating further intraorganelle electroneutral acidification (reviewed by Bradbury 1999). The chloride channels that participate in this process have not yet been identified but the results of this study suggest that mCLCA3 may be involved. It is conceivable that mCLCA3 plays a regulatory role in this pathway, similar to the central role of chloride channels in the secretion of water across epithelial membranes (Barrett and Keely 2000). This notion is supported by the previous studies of Naka-nishi and co-workers (2001) and Zhou and co-workers (2001), who convincingly demonstrated the regulatory significance of mCLCA3 in mucin secretion. In addition to its direct association with the granule membranes, mCLCA3 was also detected in the electron-dense material that buds from the luminal surface of the granule membranes during granule fusion and maturation. Although precise composition and function of this material are unknown, it has been suggested that it represents excess membrane proteins shed during fusion of the goblet granules (Swift and Mukherjee 1978; Kurosumi et al. 1981; Wille 1990).
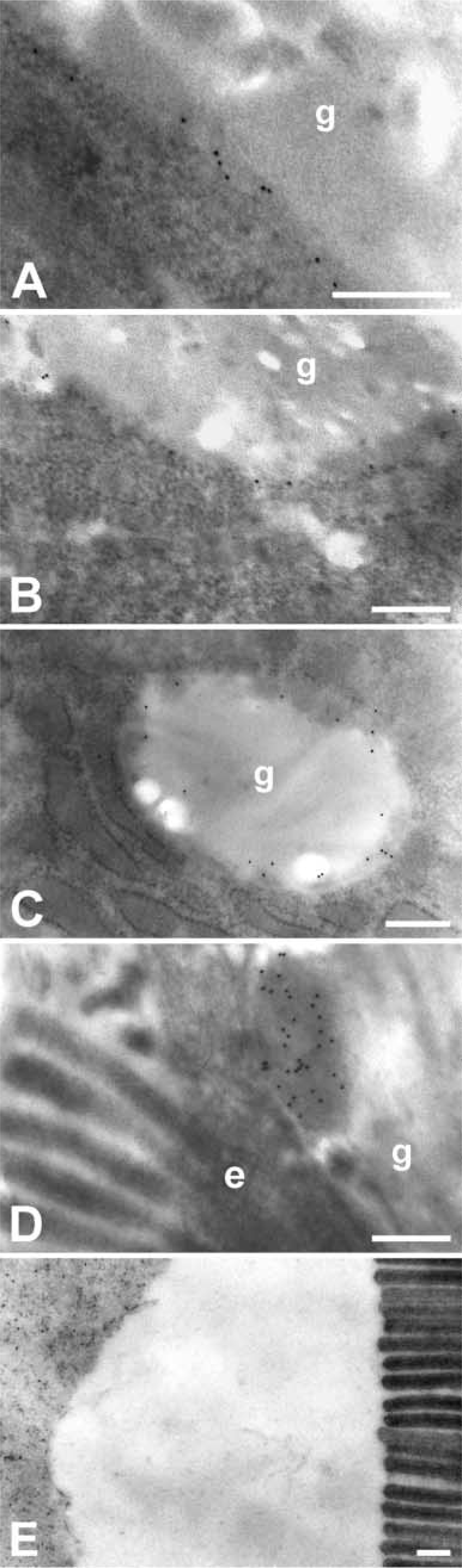
Ultrastructural localization of the mCLCA3 protein in intestinal goblet cells as detected by immune transmission electron microscopy using glutaraldehyde-formalin-fixed, LR White-embedded ultrathin tissue sections from murine jejunum (
The fusion of small granules into fewer larger granules generates excess membrane surface relative to the granule content, requiring degradation, recycling, or secretion of the superfluous material. Localization of mCLCA3 in the electron-dense material of the mucin granules, as caps on top of goblet cells, and occasionally in electron-dense regions of the intestinal luminal mucous layer suggests that it is, at least in part, released with the mucins.
Surprisingly, mCLCA3 was undetectable in specific subsets of mucin-producing cells, including those within the mucinous salivary glands and goblet cells in the base of small and large intestinal crypts. Therefore, it appears that mCLCA3 is not required for mucus secretion in general. No other murine CLCA homologues have been identified in mucin-producing cells to date, suggesting that mucin secretion may be accomplished via different pathways in different cell types. As a possible explanation for the presence of mCLCA3-positive and -negative goblet cell populations in the intestine, mCLCA3 expression may be related to the fate of the cells along the crypt-villous axis. The mitotically active goblet cell precursors are located at various positions within the intestinal crypts, giving rise to a goblet cell population that migrates to the luminal extrusion zone and sloughs off, and to a second population that migrates to the base of the crypts and undergoes apoptosis (reviewed by Colony 1996). This study shows that mCLCA3 is expressed principally in the upwardly but not in the downwardly migrating subpopulation of goblet cells in the intestine. Similarly, the gastric surface mucous cells that migrate to the luminal surface (Colony 1996) express mCLCA3, whereas those cells that migrate downwards from the mitotically active cells at the base of the gastric pits do not. Therefore, throughout the entire gastrointestinal tract, mCLCA3 appears to be expressed only in those goblet cells that migrate upwards to the extrusion zone. A functional link between CLCA homologues and apoptosis has already been observed for mCLCA1 and mCLCA2, which both appear to be involved in the induction or promotion of apoptosis (Elble and Pauli 2001).
The mCLCA3 protein is the first among members of the CLCA family to be immunolocalized ultrastructurally. On the light microscopic level, the protein expression pattern has been established only for the two bovine homologues, Lu-ECAM-1 (alias bCLCA2) and bCLCA1 (alias CaCC). Lu-ECAM-1 is a luminal membrane protein of pulmonary microvascular endothelial cells, where it is most likely involved in processes different from those of mCLCA3 (Zhu and Pauli 1991; Elble et al. 1997). In contrast, bCLCA1 is expressed exclusively on the brush border of ciliated tracheal epithelial cells, but not in goblet cells or submucosal glands (Ran and Benos 1992; Cunningham et al. 1995; Elble et al. 1997). Using in situ hybridization, the murine mCLCA1 has also been detected in ciliated tracheal epithelial cells but not in goblet cells (Gruber et al. 1998b), demonstrating that different CLCA homologues may be present in different cell types and different cellular structures within the same organ.
The cellular and intracellular expression patterns of the mCLCA3 protein are consistent with its proposed role in disorders with malfunctioning mucin secretion including asthma (as discussed above; see Nakanishi et al. 2001; Zhou et al. 2001) and cystic fibrosis (CF). CF mice suffer from severe intestinal goblet cell hyperplasia, with overproduction of a stringy mucus (for review see Grubb and Boucher 1999). Moreover, in addition to increased numbers of goblet cells and increased overall mucin production, alterations in the composition of mucins from the gastrointestinal (Clamp and Gough 1979; Wesley et al. 1983) and respiratory tracts (Frates et al. 1983; Roomans et al. 1986) of CF patients have been reported. Interestingly, the CFTR chloride channel, which is defective in CF patients, has also been localized in membranous compartments within intestinal goblet cells (Hoogeveen et al. 1991; Kalin et al. 1999), suggesting that both CFTR and mCLCA3 may be involved in mucin secretion. Clarke and co-workers (1994) postulated that a calcium-activated chloride conductance may compensate for a loss of CFTR channel function in several tissues, including the lung and intestine. Consistently, an important role of mCLCA3 in the modulation of CF disease severity has recently been proposed by Chung and co-workers (2001). In this study, mCLCA3 (alias gob-5) mRNA was found to be significantly elevated in BALB/c CF mice with a mild lung phenotype compared to C57BL/6 CF mice that exhibit severe lung pathology or compared to wild-type mice. The authors speculate that mCLCA3 expression is induced by the loss of CFTR function in BALB/c mice but not in C57BL/6 mice, and that mCLCA3 compensates for the loss of CFTR function in the airways of CF mice. The results of our study provide evidence of how mCLCA3 may be involved in this process. Further functional analyses are required to establish the role of mCLCA3 in mucous synthesis and secretion, its role in the transepithelial flux of chloride, and the signal transduction pathways that regulate the putative channel protein.
Footnotes
Acknowledgments
Supported by German Research Council (Deutsche Forschungsgemeinschaft) grants GR 1491/2-1 and GR 1491/2-2.
The excellent technical assistance of Kaethe Franke, Kerstin Rohn, and Klaus Kuhlmann is gratefully acknowledged.