
Abstract
In postnatal muscle, skeletal muscle precursors (myoblasts) can be derived from satellite cells (reserve cells located on the surface of mature myofibers) or from cells lying beyond the myofiber, e.g., interstitial connective tissue or bone marrow. Both of these classes of cells may have stem cell properties. In addition, the heretical idea that post-mitotic myonuclei lying within mature myofibers might be able to re-form myoblasts or stem cells is examined and related to recent observations for similar post-mitotic cardiomyocytes. In adult hearts (which previously were not considered capable of repair), the role of replicating endogenous cardiomyocytes and the recruitment of other (stem) cells into cardiomyocytes for new cardiac muscle formation has recently attracted much attention. The relative contribution of these various sources of precursor cells in postnatal muscles and the factors that may enhance stem cell participation in the formation of new skeletal and cardiac muscle in vivo are the focus of this review. We concluded that, although many endogenous cell types can be converted to skeletal muscle, the contribution of non-myogenic cells to the formation of new postnatal skeletal muscle in vivo appears to be negligible. Whether the recruitment of such cells to the myogenic lineage can be significantly enhanced by specific inducers and the appropriate microenvironment is a current topic of intense interest. However, dermal fibroblasts appear promising as a realistic alternative source of exogenous myoblasts for transplantation purposes. For heart muscle, experiments showing the participation of bone marrow-derived stem cells and endothelial cells in the repair of damaged cardiac muscle are encouraging.
Skeletal Muscle
Skeletal Muscle Precursor Cells and Regeneration
There is considerable interest in skeletal muscle regeneration for increased efficiency of repair in sports medicine, after severe injury or muscle transplantation, in muscular dystrophies, for possible ablation of mitochondrial myopathies, and for recovery of strength in disuse atrophy or space flight (reviewed in Grounds 1999). In all of these situations, the process of new muscle formation requires that quiescent mononucleated muscle precursor cells (myoblasts) become activated, proliferate, differentiate, and fuse together to form multinucleated young muscle cells called myotubes, and these myotubes then undergo further differentiation and (when innervated) mature to form fully functional muscle fibers (Figure 1).
Clinical Applications of Myoblast Transplantation
Beyond their role in regeneration, normal skeletal myo-blasts have been isolated, cultured, and then transplanted in vivo to replace defective genes in myopathies [e.g., dystrophin in Duchenne muscular dystrophy (DMD); Partridge et al. 1998]. The clinical possibilities of myoblast transfer therapy (MTT) have stimulated much research, although many fundamental immune problems remain to be solved (Skuk and Tremblay 2000; Smythe et al. 2001,2000b). Stem cells as an alternative source of myoblasts have particular clinical merit in the potential treatment of DMD patients by an ex vivo gene therapy approach (Floyd et al. 1998; Partridge 1998), because use of autologous cells avoids potential problems of immune rejection. Because satellite cells from the skeletal muscle of DMD boys probably have a limited capacity for replication, an alternative source of autologous myogenic stem cells (e.g., from dermal fibroblasts or bone marrow stem cells) is ideal for genetic correction and re-implantation into defective dystrophic muscles.
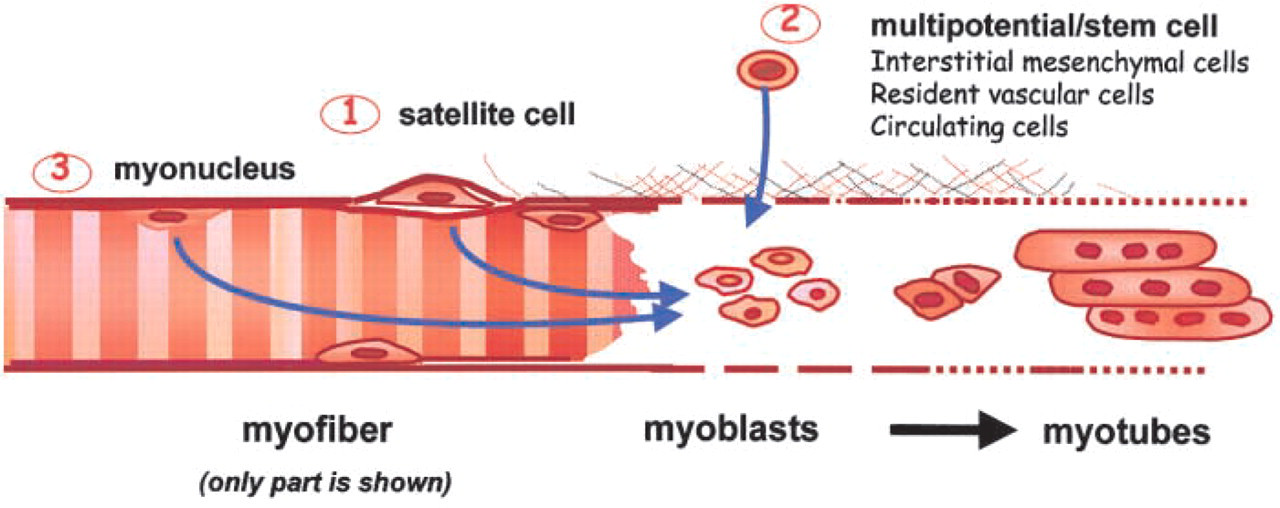
Alternative sources of myo-blasts in skeletal muscle tissue.
Transplantation of cultured myoblasts has also been used to replace defective muscles, e.g., for urinary incontinence (Yokoyama et al. 2000) and in acutely injured myocardium (Robinson et al. 1996; Dorfman et al. 1998; Hutcheson et al. 2000), and has been widely used to deliver genes into the bloodstream (Fewell et al. 2001; Lucas and Heller 2001), brain, or joints (reviewed in Grounds 1999). Cultured myoblasts are also required for the rapidly emerging discipline of tissue engineering to construct ex vivo potential artificial muscles for transplantation purposes (Vacanti and Langer 1992; Okano and Matsuda 1998; Vandenburgh et al. 1999; Grounds 2000; Wright et al. 2001). For all of these purposes, stem cells represent a potential powerful alternative source of myogenic precursor cells.
The focus of this review is the source of skeletal muscle precursor cells, and three candidate sources are considered: (a) the satellite cells of myofibers, (b) various types of cells originating outside the myofiber, and (c) “post-mitotic” myonuclei within the sarcoplasm of damaged myofibers that might re-enter the cell cycle (summarized in Figure 1). The reader is also directed to additional recent reviews of developmental relationships, therapeutic possibilities, and other aspects of skeletal muscle satellite and stem cells (Bianco and Cossu 1999; Miller et al. 1999; Cossu and Mavilio 2000; Seale and Rudnicki 2000; Bailey et al. 2001; Blau et al. 2001; Hawke and Garry 2001; Zammit and Beauchamp 2001).
Satellite Cells
Markers For Identification of Satellite Cells
Myoblasts in postnatal muscle are classically considered to be derived from cells located on the surface of the myofiber and lying beneath the basement membrane. These were originally defined on the basis of their geography and were therefore termed satellite cells. The satellite cells appear to be reserve muscle precursor cells. In mature skeletal muscles (e.g., of the limbs) they are normally quiescent and are activated only in response to growth or muscle damage. The most reliable way to identify satellite cells is on the basis of their position using electron microscopy, although this method is not particularly convenient. Therefore, much interest has focused on good antibodies to identify specific proteins in quiescent (and activated) satellite cells in vivo at the light microscopic level (Hawke and Garry 2001; Zammit and Beau-champ 2001). The main proteins identified to date as potential markers are outlined below.
Cell Surface Proteins. One of the most useful markers appears to be the cell surface protein M-cadherin (M-cad) located at the interface of the satellite cell and underlying myofiber (reviewed in Grounds 1999). Antibody detection of syndecan-3 and syndecan-4, proteoglycans that bind many growth factors, also appears to be a very promising approach to identification of quiescent and activated satellite cells (Cornelison et al. 2001). However, the rapid (but transient) increase in expression of syndecan-4 by damaged vascular smooth muscle cells in vivo (Cizmeci-Smith et al. 1997) suggests potential problems with nonspecificity with this marker for identification of satellite cells in some experimental situations with damaged muscle in vivo. Other cell surface markers for quiescent satellite cells such as c-met, the receptor for hepatocyte growth factor, are somewhat more controversial because fibroblasts and other cells in tissue sections may also express c-met (Grounds 1999).
A wide range of cell surface markers are routinely used for fluorescent activated cell sorting (FACS) analysis of hematopoetic (HSC), mesenchymal, and other stem cells (including a small side population, termed SP cells, that exclude Hoechst stain) (Thomson et al. 1998; Gussoni et al. 1999; Pittenger et al. 1999; Young et al. 1999,2001b; Beauchamp et al. 2000; Jackson et al. 2001; Reyes and Verfaillie 2001), and these markers have recently been investigated extensively on satellite cells. FACS enables separation of populations of cells that can then be studied for their lineage potentials. The simplest distinction is that all HSC are CD45+ and CD34+, whereas mesenchymal stem cells are CD45— and CD34— (Reyes and Verfaillie 2001). Myogenic cells are CD45— and most are CD34+ (Beauchamp et al. 2000), although CD34 is not a useful marker for satellite cells on tissue sections because CD34 is also present on many cells of the vasculature and on HSC (discussed in Zammit and Beau-champ 2001). Another marker of interest is Sca1 (Jankowski et al. 2001), which is present on multipotential stem cells and vascular cells but apparently not on satellite cells, although this is also debated (discussed in Zammit and Beauchamp 2001). The conflicting data are probably due in part to parameters used for FACS analysis in different laboratories, to species differences, and to possible changes in expression by cells under different conditions. Although such cell surface markers are powerful tools they also have limitations. Furthermore, they are not ideal for identifying satellite cells in vivo on tissue sections because of their expression by stem cells and other cell types that may be present in muscle tissue.
Transcription Factors. Expression of specific transcription factors is also a potentially useful marker for satellite cells. Myocyte nuclear factor (MNF) is present in quiescent and proliferating satellite cells and appears to be required for satellite cell function (Garry et al. 1997; discussed in Miller et al. 1999), suggesting that antibodies against specific isoforms of MNF (Hawke and Garry 2001) might be useful to identify such cells in vivo, although this has not been demonstrated to date.
The important observation that expression of Pax7 is essential for satellite cell formation (Seale et al. 2000) strongly suggests that isoforms of Pax 7 might be useful markers for satellite cells (Rodger et al. 1999), although Pax 7 isoforms are also expressed in other cell types (Ziman et al. 2001). Pax7 expression, as detected with immunocytochemistry (Seale et al. 2000; Zammit and Beauchamp 2001) or in situ hybridization (Seale et al. 2000) identifies candidate quiescent satellite cells in tissue sections, and it will be interesting to determine how useful Pax 7 is as a reliable marker for these cells.
The skeletal muscle-specific transcription factors MyoD and Myf5 have been intensively studied because these are key factors for acquisition of myogenic identity (Buckingham 2001) and are rapidly upregulated in activated satellite cells/myoblasts (Arnold and Winter 1998; Musaro and Rosenthal 1999; Thaloor et al. 1999). However, there are conflicting data concerning the extent to which one or both of these genes may be expressed in quiescent satellite cells and if different patterns of expression in individual myoblasts might define two populations of satellite cells (Anderson et al. 1998; Beauchamp et al. 2000; Sabourin et al. 1999). Whether Myf5-positive cells might represent a stem cell subpopulation of satellite cells (Yablonka—Reuveni et al. 1999a, b; Seale and Rudnicki 2000) also remains unclear. Alternatively, the absence of Myf5 and CD34 has been suggested as a criterion to identify a subpopulation of satellite cells that may represent stem cells (Beauchamp et al. 2000). It should be noted that markers that identify satellite cells do not address the early identification of myogenic cells derived from non-myogenic sources, and this is a separate quest (see below).
Stem Cell Subpopulation of Satellite Cells?
It has been proposed that satellite cells contain a subpopulation of cells with stem-like characteristics that serve to replenish the satellite cells' compartment, and this is the subject of an excellent recent review (Zammit and Beauchamp 2001), although there is no hard evidence to confirm this. Although heterogeneity of satellite cells (Beauchamp et al. 1999,2000; Qu et al. 1998; Edom—Vovard et al. 1999; Gross et al. 1999; Yablonka-Reuveni et al. 1999b; Jankowski et al. 2001) and immortalized myoblast cell lines (Yoshida et al. 1998) is well documented, it is not clear whether these differences reflect distinct subpopulations of cells or represent stochastic differences within one population (Zammit and Beauchamp 2001).
In support of a myogenic stem cell in muscle, an exceptional capacity for high clonal expansion of a myogenic precursor is evidenced by many dystrophin-positive myofibers generated from a single revertant nucleus in mdx muscle in vivo (Partridge unpublished observations; Zammit and Beauchamp 2001), and also by potent myogenesis of some cells derived in vitro from late passaged cells (Qu et al. 1998). However, there is no evidence to prove that the initial precursor (stem) cell in these situations (especially when cells are extracted from skeletal muscle tissue) was actually derived from a satellite cell. Although putative myogenic stem cells might developmentally arise as a subpopulation of satellite cells, they might instead originate from stem cells lying outside the muscle fiber, and these might even migrate into the satellite cell position beneath the basement membrane of myofibers. Of interest is the idea that very quiescent cells, which are slow to attach in tissue culture after cell extraction and are slow to initiate cell replication (Qu et al. 1998; Beauchamp et al. 1999), may indeed represent the most potent type of myogenic stem cell. Such a subpopulation of superior donor myoblasts has major implications as a prime source of myoblasts for transplantation purposes. That there is indeed a direct relationship between the extent of dormancy and the potency of a stem cell response is supported by classical studies of the hematopoetic lineage (Bradford et al. 1997).
The surprising observation that cells extracted from skeletal muscle, using methods used to isolate and culture satellite cells, are remarkably effective at generating cells of the hematopoetic lineages (discussed below) initially suggested a potent multipotential capacity of satellite cells. However, it appeared equally likely that such lineage plasticity could be accounted for by contaminating stem cells in the interstitial connective tissue (of non-muscle origin) extracted by the same procedure. Indeed, there is now strong evidence for (at least) two distinct lines of stem cells extracted from skeletal muscle tissue (McKinney-Freeman et al. 2002). To prove true plasticity of a satellite cell it appears necessary to grow isolated myofibers in culture (Beauchamp et al. 2000; Heslop et al. 2001), to harvest the satellite cells, and then to assess the capacity of clones to give rise to cells of different lineages. Even in this situation contaminant adherent cells, such as capillaries (Zammit and Beauchamp 2001), may complicate interpretation of such experiments.
In part this approach has been used to demonstrate that satellite cells on isolated myofibers in culture can also give rise to adipocytes or osteogenic cells (Asakura et al. 2001), although clonal cells were not tested. It is noteworthy that intrinsic lineage plasticity using isolated myofibers occurred in the absence of exogenous inducers that are required to demonstrate plasticity with cultured myoblasts (Zammit and Beau-champ 2001). Similar studies of isolated myofibers grown at different oxygen concentrations, using co-localization of markers for myoblasts and adipocytes within individual satellite cells, also strongly support the idea that satellite cells can give rise to cells of the adipogenic lineage (Csete et al. 2001). Although such observations certainly show lineage plasticity, they do not provide any insight into the existence of true stem cells with the combined capacity for extended self-renewal and plasticity within the satellite cell population.
Problems with Survival of Transplanted Cells and the Host Immune Response
For all cell transplantation therapies the introduced donor cells must be able to survive in their host environment. However, IM injection of cultured isolated (congenic) myoblasts in classical myoblast transfer therapy (MTT) shows that there is a massive and rapid necrosis of donor myoblasts, with over 90% dead within the first hour after injection (reviewed in Beauchamp et al. 1999; Hodgetts et al. 2000; Skuk and Tremblay 2000; Smythe et al. 2000b,2001). This rapid myoblast death appears to be due to exposure to tissue culture conditions (Smythe and Grounds 2000) that alter the myoblasts so that, when transferred in vivo, they provoke an acute adverse host immune response. [Note: when equivalent intact donor muscles are implanted directly, without exposure to cell isolation or tissue culture conditions, there is no adverse immune response and grafts show excellent survival for up to a year (Fan et al. 1996; Smythe and Grounds 2000)]. Host natural killer (NK) cells appear to play a particularly important role in this rapid death of cultured donor myoblasts (Hodgetts et al. 2000). The extent to which such immune problems also occur with IM transplantation of donor stem cells or with delivery of donor (stem or other) cells via the circulation is unclear (Grounds 1999), but this aspect should be carefully considered. The report that mesenchymal stem cells (MSCs) from adult human bone marrow are exempt from rejection after xenotransplantation into sheep (in contrast to other stem cells) is intriguing (Liechty et al. 2000). These donor MSC cells were detected in many tissues (although their survival was not quantitated), and such unexpected immune tolerance has major implications for clinical cell therapies and requires further investigation.
Massive death of injected donor cells is also recognized as a major problem with transplanted cardiomyocytes, especially in the inflammatory conditions that follow infarction (Zhang et al. 2001). Many cardiomyocytes die by apoptosis, and exposure of cultured cardiomyocytes to heat shock before transplantation strikingly increased cell survival at 1 day (Zhang et al. 2001). As for transplanted cultured skeletal myoblasts (Hodgetts et al. 2000), a central role for host NK cells is also implicated in cardiac allograft rejection (Maier et al. 2001), and it appears likely that the immune problems encountered with MTT will also apply to cultured cells transferred into the heart.
Overall, although it is known that satellite cells give rise to myoblasts and result in formation of new skeletal muscle, the presence of a subpopulation of satellite cells that represents a true stem cell has yet to be demonstrated. While tissue culture studies enforce the idea of plasticity between myogenic, adipogenic, and osteogenic lineages, wider lineage plasticity of a true satellite (stem) cell remains to be proved. Although cells extracted from skeletal muscle clearly show stem cell properties and considerable lineage plasticity (as discussed below), these stem cells may not be derived from satellite cells but from other cells in the tissue.
Myoblasts Originating from Cells Outside the Myofiber
The definition of stem cells is well reviewed elsewhere (Marshak et al. 2001), and it is generally agreed that stem cells have properties of plasticity and the capacity for self-renewal (i.e., cell replication). These are clearly properties of embryonic stem cells. However, do both of these properties equally apply to multipotential cells derived from adult tissues? Much recent attention has focused on aspects of plasticity, but the demonstration of plasticity provides little information about the capacity of these novel cells to undergo extensive cell replication. Indeed, the relatively low contribution of apparent stem cells derived from adult tissues to new tissue formation might (in part) reflect their limited self-renewal. There is little information available on this aspect. Although satellite cells are classically considered the source of myoblasts in postnatal muscle, the past 4 years have provided overwhelming evidence that myoblasts can also arise from non-myogenic sources in vivo (Table 1). Many of these are available within skeletal muscle tissue (Figure 1), whereas others are exogenous and therefore are unlikely to routinely contribute to skeletal muscle formation. It should be noted that few of these plastic cells, would probably qualify as stem cells. There are clinical applications for such alternative sources of myoblasts and for putative myogenic stem cells, and this is now a topic of intense research interest. The many potential sources of such myogenic cells originating beyond the myofiber are outlined below.
Resident Cells in Non-muscle Tissues That Give Rise to Myoblasts
Ready Interconversion of Mesenchymal Cells. Interstitial connective tissue contains many cell types. There is a plasticity of mesenchymal cells (in interstitial connective tissue) and interconversion among myoblasts, fibroblasts, adipocytes, chondroblasts, and osteoblasts has long been recognized and can be demonstrated in tissue culture under different conditions, as indicated in Table 2 (see also Asakura et al. 2001; Zammit and Beauchamp 2001). In most cases, specific inducers are required for conversion into the different lineage in vitro. This raises the question of the extent to which such inducers or precise conditions might exist in vivo to produce lineage switching. The role of inducers is discussed below in more detail. Although desmin is a very useful marker for muscle cells, it is expressed by all muscle lineages, including skeletal, cardiac, and smooth muscle (visceral and vascular). Therefore, confirmation of the presence of skeletal muscle cells specifically requires the additional expression of skeletal muscle-specific markers such as the transcription factors MyoD, Myf5, or myogenin, specific actins or myosins, or the formation of multinucleated myotubes. Fibroblasts appear to be particularly inclined to convert into myoblasts. This is demonstrated by forced expression of the skeletal muscle-specific transcription factor MyoD in fibroblasts and other cell lines (after transfection with cDNA for MyoD), where fibroblasts showed the greatest propensity for myogenic conversion (Weintraub et al. 1991; see also Table II in Walker et al. 2001). A greater efficiency of conversion was shown with viral delivery of MyoD, and dermal fibroblasts were better (60–90% myogenic conversion) than fibroblasts derived from skeletal muscle of mice and humans. Furthermore, these converted fibroblasts differentiated, fused, and formed skeletal muscle in vivo (Lattanzi et al. 1998). Dermal fibroblasts can also be converted into (desmin-positive) myoblasts by co-culture with skeletal muscle (see below).
Plasticity: summary of different cell types of non-muscle origin that give rise to skeletal and cardiac muscle cells a
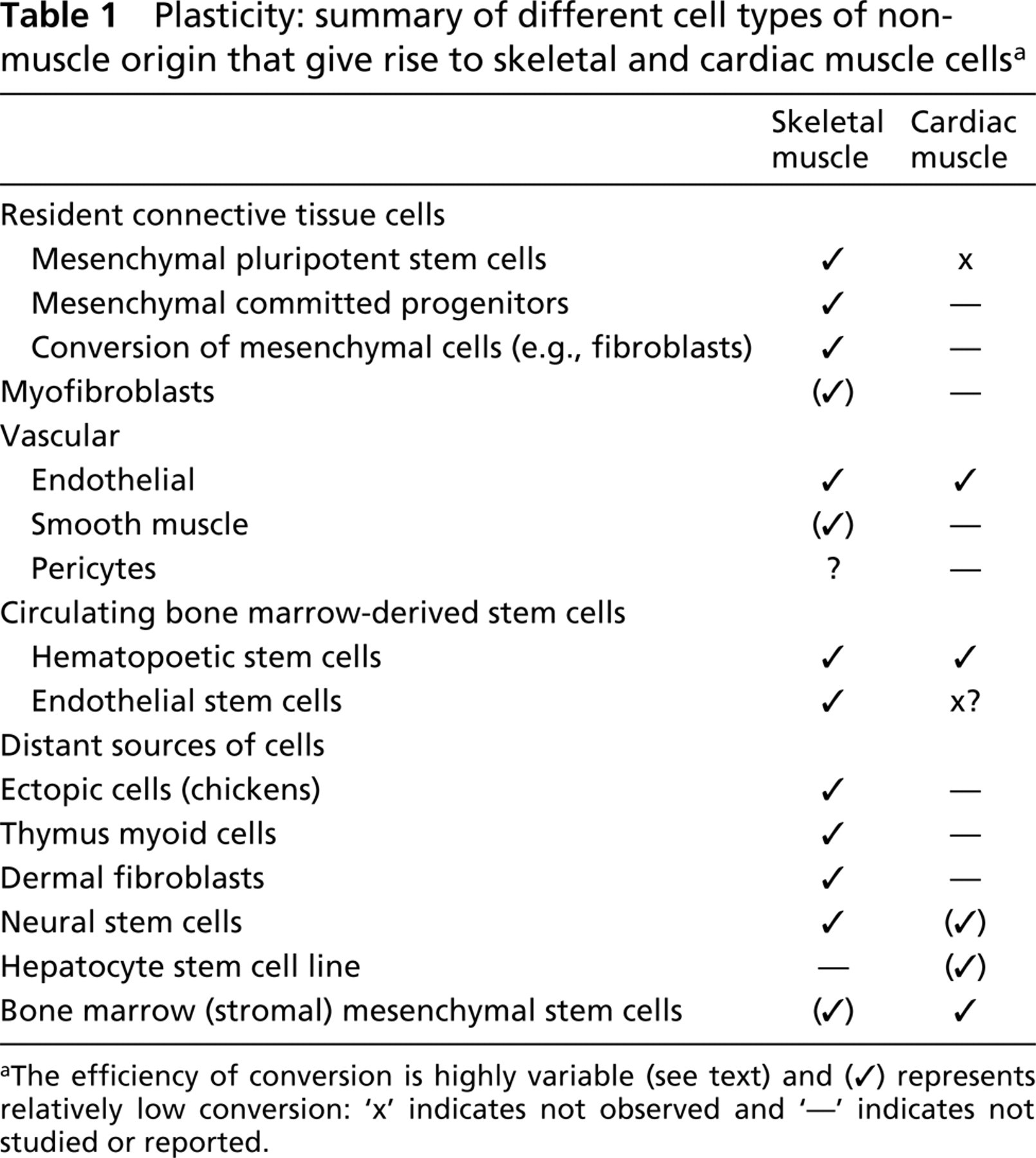
aThe efficiency of conversion is highly variable (see text) and (✓) represents relatively low conversion: ‘x’ indicates not observed and ‘— indicates not studied or reported.
Mesenchymal Stem Cells. At least two populations of mesenchymal stem cells (MSCs) that can be extracted from connective tissue of many species have been described extensively by Young and colleagues (Young et al. 1998,1999,2001a, b). These are progenitor cells (with restricted but committed lineages) and pluripotent cells (with no committed lineage). Both types of stem cells are present in fetal, adult, and geriatric human dermis and skeletal muscle (Young et al. 2001b). Because MSCs are also derived from the bone marrow throughout life (Reyes and Verfaillie 2001), it is difficult to distinguish between the possibilities that pluripotential cells in the interstitial connective tissue are indeed derived from endogenous resident cells or that they might originate from circulating bone marrow-derived precursor cells, especially during development. It is noted that although these pluripotential MSCs derived from dermis and skeletal muscle tissue do give rise to skeletal muscle cells (which express myogenin, sarcomeric myosins, and fuse to form myo-tubes) under the influence of dexamethasone and insulin, they do not form cardiac muscle cells (Young et al. 2001a). In contrast, bone marrow-derived MSCs appear to readily give rise to cardiac muscle (discussed below).
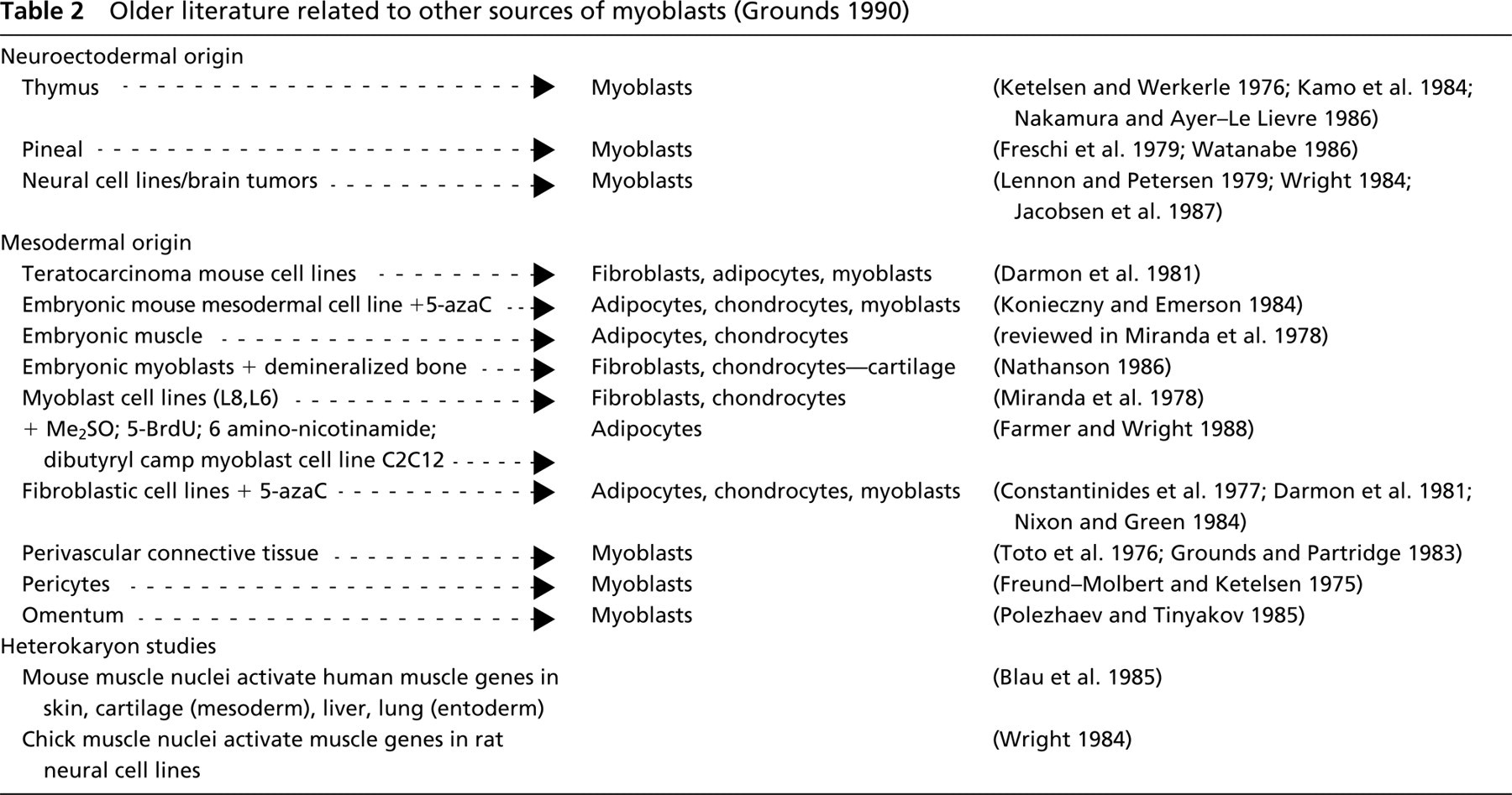
Older literature related to other sources of myoblasts (Grounds 1990)
Dermal Fibroblasts and Stem Cells. The ability of dermal fibroblasts to fuse spontaneously with developing myotubes was demonstrated for dysgenic muscle and resulted in genetic and functional rescue of the defective muscle (Courbin et al. 1989; Chaudhari and Beam 1990). Many studies have now shown that dermal fibroblasts readily convert into myoblasts (which express a range of skeletal muscle specific markers and fuse to form myotubes) when co-cultured with skeletal muscle in vitro (Breton et al. 1995; Salvatori et al. 1995; Wise et al. 1996), and they can also contribute to new skeletal muscle fibers in vivo (Gibson et al. 1995). The observation that conversion to myoblasts occurs only with dermal but not skeletal fibroblasts (Wise et al. 1996; Goldring et al. 2002b) favors the idea of a special class of fibroblasts found only in the dermis and not in skeletal muscle. It is not clear whether these fibroblasts represent a stem cell. An alternative explanation for the lack of such plastic fibroblasts in skeletal muscle is that the same candidate population has already been converted into myoblasts in the myogenic environment.
There may be several kinds of stem cells resident in the dermis with different lineage capabilities (Slack 2001; Toma et al. 2001). The non-adherent skin-derived precursors isolated from both rats and humans that give rise to both mesodermal (adipocytes and smooth muscle cells) and neural lineages (Toma et al. 2001) appear to be different from the various classes of mesenchymal stem cells described by Young (Young et al. 1994,2001b) and from the adherent cells of Watt (Goldring et al. 2000). The impressive conversion of mouse and human dermal fibroblasts to desmin-positive cells in vitro (when co-cultured with skeletal muscle cells or in the presence of galectin-1) (Goldring et al. 2002a) raises real possibilities for the use of dermal cells as an alternative source of myo-blasts, especially for ex vivo gene correction of autologous cells for cell transfer therapy (Partridge 1991a), and this source is particularly attractive for clinical applications.
Myofibroblasts. Myofibroblasts are classically considered to be intermediate between fibroblasts and smooth muscle cells (reviewed in Powell et al. 1999; Walker et al. 2001). Myofibroblasts resemble fibroblasts but ultrastructurally have some features of muscle. They are present in almost all organs and are associated with wound repair and fibrosis. The origin of these cells is unclear. They may be derived from fibroblasts or may correspond to pericytes closely associated with blood vessels, and they can be derived from embryonic stem cells in the presence of PDGF and stem cell factor (Powell et al. 1999; Walker et al. 2001). Myofibroblasts are usually identified by expression of the cytoskeletal protein desmin (which is present in smooth, skeletal, and cardiac muscle), often in addition to vimentin (which is a marker for fibroblasts) and by smooth muscle α-actin, but they do not express cardiac or skeletal α-actin (Powell et al. 1999). Expression of the skeletal muscle-specific genes for MyoD, myogenin, and sarcomeric myosins in myofibroblasts in culture and also in rat liver stellate cells in vivo confirms that they also have some properties of skeletal muscle (Mayer and Leinwand 1997; Walker et al. 2001). However, myofibroblasts do not appear to express Myf5, to terminally differentiate, or to fuse to form myotubes in culture (Walker et al. 2001), although the possibility that such a transition might occur in damaged tissues in vivo cannot be excluded. These observations raise interesting questions about the precise status of these connective tissue cells and the molecular control of myogenesis (Walker et al. 2001).
Vascular Tissue. Similarly, lines of vascular smooth muscle cells in culture have been shown to express MyoD, myogenin, and several skeletal muscle-specific structural genes, but it should be noted that they do not express Myf5 and only some cell lines could fuse to form myotubes (Graves and Yablonka Reuveni 2000). Although transitions from smooth to skeletal muscle in vivo have been reported, these have been difficult to confirm with available markers (Graves and Yablonka Reuveni 2000). It now appears that the close association between smooth and skeletal muscle cells in the fetal esophagus may reflect different populations of precursor cells rather than products of trans-differentiation (Zhao and Dhoot 2000). Heterogeneity of gene expression in vascular smooth muscle cells in distinct vascular beds was demonstrated by studies with LacZ-marked transgenic mice, and one transgene expressed in pericytes in adult mice was also expressed during development in migrating myoblasts (and also in thymic epithelium) (Tidhar et al. 2001). In the vascular system, there is a close relationship and possible trans-differentiation among vascular smooth muscle cells, pericytes, and endothelial cells (DeRuiter et al. 1997). The nature of pericytes is unclear, but it was proposed almost 30 years ago that pericytes could give rise to skeletal muscle (see Table 1).
There is now strong in vivo evidence that skeletal muscle precursors can arise from cells in the vasculature (reviewed Bianco and Cossu 1999; De Angelis et al. 1999), although the precise origin of these precursors is unclear (discussed in Zammit and Beauchamp 2001). The cells in the embryonic dorsal aorta that give rise to myoblasts may be related to endothelial cells and of bone marrow origin (Bianco and Cossu 1999; De Angelis et al. 1999). Trans-differentiation of endothelial cells into cardiomyocytes has also been demonstrated (Condorelli et al. 2001). Tissue structure in vivo is complex, and the extent to which such vascular cells (endothelium, smooth muscle and peri-cytes) may contribute to the various stem cell populations extracted from various tissues must be considered.
Neural Tissue. That neural tissue can give rise to skeletal muscle cells (including myotubes) has long been recognized (Table 1; and Tajbakhsh et al. 1994), and recent data implicate a resident multipotential stem cell in neural tissue. During development it has been shown that Myf5-positive cells are present in embryonic mouse neural tube tissue. These neuroectodermal cells co-express neural and muscle markers and can form mononucleated skeletal muscle cells in culture (Tajbakhsh et al. 1994). Studies confirming that neuron-derived clonal cells can give rise to skeletal muscle in culture (Galli et al. 2000; Rietze et al. 2001) emphasize that cell contact with C2C12 or primary myoblasts was required for conversion of the neural stem cell into the myogenic lineage (manifested by differentiated myoblasts or fusion to form myotubes), and it was also observed that fusion into myotubes occurred without replication of the neural stem cell (Rietze et al. 2001). These neural stem cells can also give rise to heart muscle, but with a lower efficiency (Condorelli et al. 2001). Whether such stem cells in neural tissue are possibly derived during development from bone marrow stem cells is unclear.
Mysterious Myoid Cells in the Thymus and Other Ectopic Myogenic Cells. It seems pertinent to examine the well-documented existence of skeletal muscle precursor cells in tissues (considered to be of neuroectodermal origin) such as the thymus and pineal. Although it has been known for over 30 years that skeletal myocytes are readily grown from such tissues in vitro, striated skeletal muscle cells are also formed in thymus tissue in vivo, especially in birds and reptiles and sometimes in humans (reviewed in Grounds et al. 1992). These cells in the thymus are termed myoid cells. It has been suggested that myoid cells might contribute to endogenous skeletal muscle regeneration (Wong et al. 1999), and transplantation studies show that myoid cell lines can indeed form new skeletal muscle in vivo (Pagel et al. 2000). The existence of these myoid cells raises many interesting questions about their origin and function (Grounds et al. 1992), their relationship to stem cells, and the factors that control their myogenic expression both in vivo and in vitro. Myoid cells in the thymus present a very potent opportunity for comparison with other more well-studied myogenic stem cells, and intensive parallel studies of these mysterious myoid cells might yield valuable insights into many aspects of myogenesis.
A recent report (Gerhart et al. 2001) showed that myogenic (MyoD-positive) cells were present in many tissues (brain, lung, intestine, kidney, spleen, heart, liver) in fetal chickens and that these cells readily formed skeletal muscle in culture. These interesting observations indicate a widespread distribution of such committed myogenic precursors in many organs in chickens. It may be that in other species the distribution of such cells is more restricted (or less apparent), especially in mature organs, and that the myoid cells in the thymus of mammals actually represents this equivalent population that can indeed be identified (Grounds et al. 1992). Whether similar ectopic committed skeletal muscle cells are resident in other organs in mice and men remains to be determined. An earlier stage of such ectopic myogenic cells might correspond (at least in part) to the committed muscle progenitor stem cells isolated from connective tissue of dermis and skin (Young et al. 2001b).
Circulating Bone Marrow-Derived Stem Cells
Bone marrow contains many cell types, including hematopoetic stem cells (HSCs), mesenchymal stem cells (MSCs), and endothelial stem cells (Goodell et al. 2001), and the gene regulation and relationship between these stem cells is complex (Baron 2001; Bianco et al. 2001; Hirschi and Goodell 2001; Nishikawa 2001; Reyes and Verfaillie 2001). Throughout life, the HSCs and endothelial stem cells circulate in the blood, whereas in mature animals the MSCs probably remain in the bone marrow (as the stroma to support the maintenance and expansion of other local stem cells), although it is not clear whether these MSCs might also circulate during development. The same postnatal human marrow MSCs that can form skeletal muscle (discussed in Zammit and Beauchamp 2001) are also able to give rise to endothelial cells (Reyes and Verfaillie 2001) and other cell types, and they appear to be closely related to pericytes in the marrow vasculature (Bianco et al. 2001).
To detect the contribution of circulating (donor) bone marrow cells to skeletal muscle formation in vivo, donor cells are now often identified in transplantation studies using a Y-chromosome (male)-specific probe to track donor male cells in female host tissues (Grounds et al. 1991; Irintchev et al. 1998; Beauchamp et al. 1999; Bittner et al. 1999; Gussoni et al. 1999; Hodgetts et al. 2000; Smythe et al. 2000a) or using genetically engineered donor cells that express a reporter gene such as Lac Z, usually driven by a muscle-specific promoter (Ferrari et al. 1999; Lescaudron et al. 1999; Torrente et al. 2000; Heslop et al. 2001).
The recent demonstration that muscle nuclei can arise from bone marrow-derived precursor cells in vivo (Ferrari et al. 1998,1999; Bittner et al. 1999; Gussoni et al. 1999) excited considerable interest for potential therapy, although this conversion appears to be a fairly rare event. Earlier bone marrow cross-transplantation studies designed to demonstrate such a relationship (Grounds 1983; Watt et al. 1987) were limited by the sensitivity of available (isoenzyme) cell markers and serve to emphasize the normally very low level of bone marrow cell contribution to skeletal muscle formation. It should be noted that the contribution of such donor myogenic cells occurred only in muscle regenerating after damage and, in many (but not all) instances, occurred in irradiated host muscle (Bittner et al. 1999; Ferrari et al. 1999; Gussoni et al. 1999), which impairs regeneration from endogenous precursors and is an unusually mitogenic environment for donor myoblasts. Assessment of the efficacy of replacement of the missing protein dystrophin by normal bone marrow-derived muscle nuclei in dystrophic muscles of mdx mice (a model for DMD) is complicated by the presence of host-revertant muscle fibers that can express endogenous dystrophin (Partridge 1991b). This complication can be avoided by using dystrophic mdx4CV mice that have very few revertant fibers. Studies using these host mice emphasize the low contribution to new muscle formation of bone marrow-derived cells because such donor cells contributed less than 1% of new myofibers over 10 months (Ferrari et al. 2001). In addition, a fortuitous human situation was recently reported by Gussoni and colleagues (unpublished data) in which a boy with DMD received a bone marrow transplant at 1 year of age and is now aged 14, but his dystrophic (exon 45 deletion) skeletal muscles showed only a very low contribution of “normal” myofibers derived from the donor bone marrow cells, even after 13 years. The precise nature of the skeletal muscle (stem) cell derived from bone marrow (i.e., HSC, MSC, endothelial, or other) remains to be defined.
Of particular interest was the demonstration of hematopoietic lineages arising from stem cells extracted from skeletal muscle (Gussoni et al. 1999; Jackson et al. 1999; Goodell et al. 2001; Kawada and Ogawa 2001). Although this was also reported for neural tissue (Bjornson et al. 1999), it has been difficult to repeat. Stem cells isolated from skeletal muscle are potent at repopulating the blood lineages (Jackson et al. 1999; Goodell et al. 2001), and the relationship of these HSCs to myogenic stem cells derived from skeletal muscle has recently been addressed. It has been shown that the HSC are present only in the Ly-5 + (CD45+) fraction of cells extracted from skeletal muscle (Kawada and Ogawa 2001), and there is now strong evidence for (at least) two separate populations of stem cells within skeletal muscle tissue: a lineage of classical HSC (CD45 +, Sca1 +) in addition to a separate lineage of myogenic precursor cells (mainly CD45—, Sca1 +) (McKinney-Freeman et al. 2002).
Bone marrow-derived stem cells can give rise to many other cell types apart from skeletal muscle, including cardiac muscle (Bittner et al. 1999; Makino et al. 1999; Tomita et al. 1999; Jackson et al. 2001; Orlic et al. 2001), hepatocytes (Petersen et al. 1999; Krause et al. 2001), neural, and other cells (Pittenger et al. 1999; Weissman 2000; Bianco et al. 2001; Goodell et al. 2001), and more examples of such broad lineage possibilities may emerge. There appears to be great plasticity of adult stem cells (Goodell 2001), and stem cells from diverse tissues may be more totipotential and possibly similar than was previously anticipated (Almeida-Porada et al. 2001; Blau et al. 2001; Zammit and Beauchamp 2001).
It is possible that circulating stem cells during development might give rise to many of the stem cells ascribed to specific tissues. In postnatal tissues, it is not clear whether bone marrow-derived stem cells contribute to cells in the vasculature or become resident in interstitial connective tissue. However, from studies of clearance of GFP-labeled lineage-negative blood cells, it has been calculated that 20,000–100,000 HSCs/progenitors enter the blood every day, and these are rapidly sequestered (within minutes) into tissues (I. Weissman 2001, personal communication). These observations on circulating bone marrow-derived stem cells have attracted great interest because delivery of muscle precursors through the bloodstream represents an ideal route for distribution to all skeletal muscles. It should be considered that the use of umbilical cord blood may be an alternative and possibly superior source of multipotential stem cells (Lewis and Verfaillie 2000), and banking of such cord cells has been proposed for future clinical applications. Because adherent MSCs obtained from adult bone marrow can expand well in culture (in contrast to HSCs), marrow stromal cells may also be a good source of therapeutic cells for transplantation purposes (Bianco et al. 2001; Reyes and Verfaillie 2001). A critical question relates to the potential scale of contribution of such non-muscle stem cells, especially those derived from adult tissues, to new muscle formation in vivo.
Inducers to Enhance Recruitment of “Stem” Cells into the Myogenic Lineage
It has long been recognized that the immediate environment can radically alter the phenotype of a cell and re-direct its gene program. Complex interactions between cells and their extracellular matrix environment clearly play a central role in determining cell fate (Prosper and Verfaillie 2001; Zamir and Geiger 2001). This has been dramatically demonstrated for inter-conversion of mesenchymal and epithelial cells (Bissell et al. 1982), and the correct interactions between cells and the bone marrow microenvironnment determine the fate of HSCs and their progenitors (Prosper and Verfaillie 2001). Similarly, the developmental fate of embryonic stem cells is determined by interactions with the local ECM (via integrins) as well as soluble factors (Czyz and Wobus 2001). Furthermore, intimate cell-cell contact is often also essential to change the fate of a cell.
Such studies have understandably often been carried out in the artificial environment of tissue culture. The particular tissue culture conditions used can dictate the cell response and the lineage pathway. This is well illustrated by changes in oxygen levels. The proliferation of old (Chakravarthy et al. 2001) and young (Csete et al. 2001) satellite cells is greatly improved in low (2–6%) oxygen levels (this is also the case for neural stem cells), and satellite cells and mesenchymal cell lines have a propensity to acquire an adipocyte phenotype when grown under high (20%) oxygen conditions traditionally used in tissue culture, compared with low physiological (2–6%) oxygen levels (Csete et al. 2001). Clearly, in vitro and in vivo conditions are vastly different. Although it is very difficult to unravel the complexity of factors and interactions present in the in vivo environment, ultimately it is the local in vivo microenvironment that dictates the behavior and fate of a particular cell. Few experiments to date have investigated conditions that influence the recruitment of stem cells into the myogenic lineage.
One powerful inductive stimulus to convert pluripotent cells into a committed lineage is dexamethasone, whereas insulin (or insulin-like growth factors-I and -II) is required to induce myogenic expression in progenitor cells. It is interesting to note that many weeks are required for this conversion in vitro (Young et al. 1998,2001a, b).
A particularly interesting factor is galectin-1, which induces conversion of primary cultures of mouse (Watt et al. 1998; Goldring et al. 2000) and human (Goldring et al. 2002a) dermal fibroblasts to myoblasts with about 30% efficiency, and 100% conversion to the myogenic lineage is seen with cloned dermal fibroblasts (Goldring et al. 2000). Whether this conversion represents trans-differentiation or a multipotential stem cell (perhaps with a wider lineage capacity) remains to be determined. The conversion of untreated dermal fibroblasts into new muscle in dystrophic mdx mice indicates that some inductive signals are normally present in degenerating/regenerating skeletal muscle in vivo (Gibson et al. 1995), and indeed galectin-1 is abundant in muscle (Goldring et al. 2002b). The nature of such inductive signals in (damaged) muscle, and the extent to which they might act on mesenchymal or other local stem cells in the interstitial connective tissue of skeletal muscle in vivo, remain to be elucidated.
Overall, it appears that the wide range of non-myogenic sources of myoblasts (Table 1) fall broadly into four classes, as outlined in Table 3. Many of these are present in skeletal muscle tissue as potential alternative sources of myoblasts, while others (e.g., dermal fibroblasts) are not. Normally there appears to be a very low contribution of these alternative myogenic precursors to muscle formation in vivo. Much work remains to be done to clarify the relationship between these various myogenic precursor cell types. The identification of inductive agents to increase recruitment of non-myogenic (stem) cells into the skeletal muscle lineage (ideally in vivo), and factors that can increase the numbers of such cells in situ are two main challenges to overcome before potential clinical application can realistically be considered.
Beyond the myofiber: four broad classes of cells derived from sources other than skeletal myofibers that can give rise to myoblasts (and form skeletal muscle)

Can Myonuclei Give Rise to Skeletal Myoblasts?
Traditionally, the answer to this question for mammalian muscle has been an unequivocal “NO.” However, this idea is rather attractive because efficient salvage of myonuclei from damaged myofibers could provide a large pool of new myoblasts for muscle repair: There are two aspects to such “recycling” of myonuclei. One is the formation of new membranes around myonuclei and fragmentation of the myotube/myofiber to form individual mononucleated cells, and the second is reinitiation of DNA synthesis in “post-mitotic” nuclei. Exciting new developments from studies of amphibian myotubes in vivo and in vitro and from tissue culture experiments with mamalian myotubes unequivocally show that reversal of myonuclear fate is possible in some situations. However, little is really known about local conditions/inducers in damaged mammalian muscle regenerating in vivo that might facilitate such events.
Amphibians
It has long been recognized that adult urodele amphibians such as the newt, have a remarkable regenerative ability for many tissues, including whole limbs (Carlson 1970b,1982; Hay 1970) and ventricular and atrial cardiac muscle (reviewed in Brockes 1997). A critical aspect of this is the ability of differentiated cells to reenter the cell cycle. For newt myotubes in tissue culture, the presence of thrombin and serum stimulates myonuclei to re-enter S-phase via phosporylation of the retinoblastoma protein (Tanaka et al. 1999). The capacity of newt myonuclei to synthesize DNA within myotubes, and also to give rise to myoblasts with high efficiency, was importantly confirmed in vivo in the blastema (mass of undifferentiated/de-differentiated proliferating cells in the growth zone under the wound epidermis formed after damage) of amputated newt limbs (Kumar et al. 2000).
Mammals
Tantalizing EM observations of vesicles accumulating around myonuclei in damaged mouse myofibers (Figure 2) support the possibility of sequestration of myonuclei by newly formed membranes to form myoblasts, but this is only suggestive and certainly is not strong evidence because there are many alternative interpretations of such static images. These images correspond to sequestration of myonuclei demonstrated in the blastema of regenerating salamander muscles (Carlson 1970b; Hay 1970). Similarly, EM observations of unusual clefts and vesicles around muscle nuclei in damaged muscles of rabbits and mice were interpreted as evidence for possible sequestration of myonuclei to form myoblasts (Reznik 1970). Such potential sequestering of myonuclei into mononucleated cells and re-initiation of DNA synthesis in new myoblasts has been vigorously debated in the past (Carlson 1970a; Pullman and Yeoh 1978) and has been exceedingly difficult to test in vivo to date.
In mouse myotubes in culture, re-initiation of DNA synthesis can occur by overexpression of the oncogenic SV40 big-T-antigen that inactivates tumor suppressor Rb and re-activates Cdc2-cyclin B, and this can lead to mitosis and cytokinesis (Iujvidin et al. 1990; Endo and Nadal-Ginard 1998). Although myonuclei in mammalian myotubes are normally refractory to stimulation by serum and thrombin (Velloso et al. 2001), studies of hybrid newt and mouse myotubes show that mouse myonuclei also respond in the presence of newt myonuclei (Velloso et al. 2001). This obervation, combined with the fact that DNA synthesis can be re-initiated by transfer of the myonucleus to oocyte cytoplasm (Wilmut et al. 1997), emphasizes the importance of cytoplasmic factors.
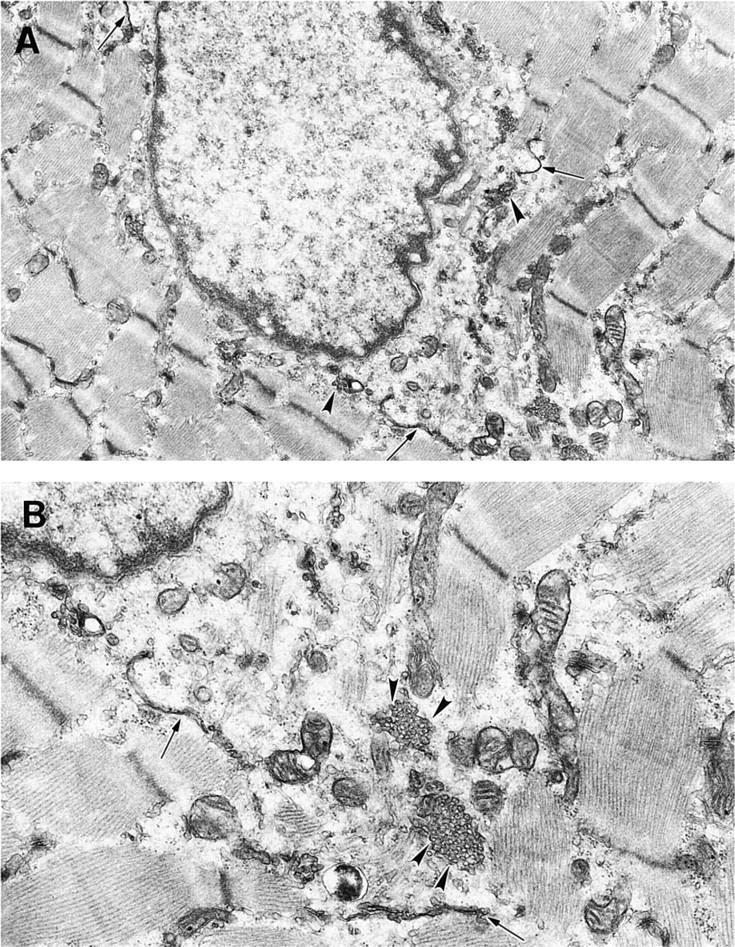
Possible formation of new membrane around a myonucleus in damaged muscle. (
Factors Involved in De-differentiation of Myonuclei
The intriguing question is, what are the in vivo factors that stimulate the de-differentiation of nuclei within myofibers and do such conditions occur in damaged mammalian skeletal muscle? Recently, information gleaned from studies of newt myogenesis has been applied to mammalian myotubes, and it appears that the two processes of DNA synthesis and fragmentation of the myotube/myofiber can operate independently (reviewed in Brockes et al. 2001). A microtubule-binding purine called myoseverin causes myoblasts to be generated from myotubes of the mouse myogenic C2C12 cell line (and also from newt myotubes) (Rosania et al. 2000; discussed in Brockes et al. 2001), and other tubulin-binding molecules (colchicine, vinblastine, taxol) similarly caused myotube disassembly. This interesting report showed that the myoblasts formed (by membrane formation around myonuclei), especially by myoseverin, can proliferate and form new myo-tubes and that myoseverin regulates transcription of many genes that influence regeneration.
Disassembly of myotubes also results from ectopic expression of the transciptional repressor protein msx1, which is implicated in de-differentiation: msx-induced cleavage of C2C12 myotubes (seen initially in about 9% of treated myotubes) resulted in the formation of either smaller myotubes or myoblasts (5.4%) capable of replication (Odelberg et al. 2000). These myoblasts could also give rise to other cell lineages, e.g., chondrogenic, adipogenic, osteogenic (Odelberg et al. 2000). This is similar to the plasticity observed with cultured satellite cells/myoblasts as discussed above (e.g., Table 2). Perhaps the expression of msx1 in quiescent satellite cells (Cornelison et al. 2000) contributes to such lineage plasticity. The observations of Odelberg et al. (2000) appear to support the idea that the mammalian myonuclei first form myoblasts and then proliferate, rather than nuclear replication within the synctium. Further studies using protein extracts from regenerating newt limbs applied to cultured C2C12 myotubes showed decreased expression of muscle differentiation proteins and maximal proliferation (BrdU incorporation) of myo-tube nuclei by 4 days (McGann et al. 2001), and careful observation confirmed cleavage by 4 days in 11% of these treated myotubes. Whether factors (such as myoseverin or msx1) that facilitate the conversion of myonuclei to myoblasts are increased in damaged mammalian skeletal muscle in vivo remains to be determined.
These fascinating observations show that the muscle nuclei in mature mammalian myotubes (like their amphibian counterparts) are capable of de-differentiating to form cells that are like myoblasts or multipotential cells, when exposed to appropriate signals. The extent to which this might normally occur in damaged skeletal muscle remains an open question. Such reversal of the post-mitotic status of myonuclei (in the highly differentiated muscle fiber) also has implications for cardiac myocytes.
Cardiac Muscle
Growth, Damage, and Repair
Hypertrophy, Polyploidy, and Multinucleation. Cardiac and skeletal muscles have a similar sophisticated organization of contractile proteins into sarcomeres and are collectively referred to as striated muscles. Growth of the heart is generally characterized by division of muscle cells during the embryonic stages of life, followed after birth by entry into a post-mitotic state. Therefore, growth of the heart during normal development and in cases of cardiac disease requires enlargement of individual cardiomyocytes (hypertrophy) rather than proliferation of post-mitotic cardiac myocytes. The average size of mature cardiomyocytes in a wide range of species appears to be 11–18 μm (Adler et al. 1996). In most species there is some additional nuclear division in the absence of mitosis, thus generating cardiomyocytes with two or more nuclei (Li et al. 1997a), although it is not clear at which stage of development this occurs. The situation in the human and other primates appears to be different because mature human cardiac myocytes are predominantly (74%) mononucleated, with smaller populations of binucleated, trinucleated, and tetranucleated cells (25.5%, 0.4%, and 0.1%, respectively) (Olivetti et al. 1996). However, humans and primates have increased ploidy, (i.e., increased numbers of copies of DNA within a single nucleus) compared with the other species (Adler et al. 1996). In humans, 20.5% of cardiomyocytes are diploid (2c) and 56.7% are tetraploid (4c) or higher, whereas 80% of nuclei are diploid in the other species. Higher ploidy (greater than 2c) indicates that DNA replication has indeed occurred within human cardiomyocytes but that the chromosomes have not separated to form two new nuclei; furthermore, this ploidy increases with age. Therefore, there appears to be a direct relationship between increased ploidy and decreased multinucleation in postnatal cardiomyocytes. These observations suggest that, after DNA replication, cell division is blocked at the stage of chromosomal separation in humans (and primates), whereas additional nuclei are generated in other species and the block to cardiomyocyte replication/division occurs later at cytokinesis. The reason for this species difference remains unclear. However, it may affect strategies to increase existing cardiomyocyte numbers in mice and humans because the block to cell replication appears to occur at an earlier stage (separation of chromosomes) in humans compared to mice.
Lack of Regeneration of Cardiac Muscle. In a normal situation, this hypertrophic growth is sufficient to maintain adequate cardiac myocyte function, but significant problems arise when the myocytes are damaged in conditions such as cardiac infarction. The postnatal capacity for cell replication during growth and regeneration of cardiac and skeletal muscle is markedly different. As outlined above, skeletal muscle cells are multinucleated and can readily regenerate from precursor cells (satellite cells/myoblasts), whereas until very recently it was established dogma that postnatal cardiac muscle was incapable of tissue repair. This was because (like skeletal myofibers) the mature cardiac cells were post-mitotic and incapable of replication (either in vivo or in vitro) but (in marked contrast to skeletal muscle) there appeared to be no reserve precursor cells. Therefore, damaged heart muscle was replaced by scar tissue, presenting a major clinical problem (Figure 3B). However, this dogma is now being replaced by the idea that there is some in vivo proliferation of cardiomyocytes after damage (see below): whether this occurs from pre-existing mature cardiomyocytes or from local stem cells is now an area of intense research.
Strategies for Repair. When confronted with the problem of heart damage after infarction, replacement of myocytes is the ideal scenario. There is increasing interest in cell transplantation as a potential therapy for cardiovascular disease (Reinlib and Field 2000). Cardiomyocyte replacement could be achieved by stimulating proliferation of endogenous mature cardiomyocytes or stem cells, or by implanting exogenous donor cardiomyocytes possibly derived from stem cells (Figure 3B).
It should be emphasized that, to be clinically effective, the enhancement of cardiomyocyte proliferation after damage must be rapid if the animal is to survive any acute insult that disrupts heart function through damage to the individual cardiac myocytes. In cases in which there is chronic damage to the heart muscle, the proliferation might need to be sustained to slowly but continually replace myocytes as necessary during the course of the disease. Whatever the source of the cells and the use to which they are put, a concurrent revas-cularization must also keep pace with repopulation of the infarct to ensure viability of the repaired region and prevent further scar tissue formation. Finally, the newly formed myocardium must integrate into the existing myocardial wall if it is to assume the function of the tissue it replaces. All of this must occur while the heart continues to beat and perform the essential work of supplying blood throughout the organism. Furthermore, even small areas of imperfectly integrated tissues are likely to severely alter electrical conduction and syncytial contraction of the heart, with long-term life-threatening consequences. This is in marked contrast to the situation in skeletal muscle where the tissue can rest during repair. Here we discuss the historical use of transplanted skeletal muscle cells and fetal cardiomyocytes to replace damaged myocardium, reinitiation of mitosis in mature “post-mitotic” cardiomyocytes, and formation of cardiomyocytes from stem cells (endogenous and exogenous).
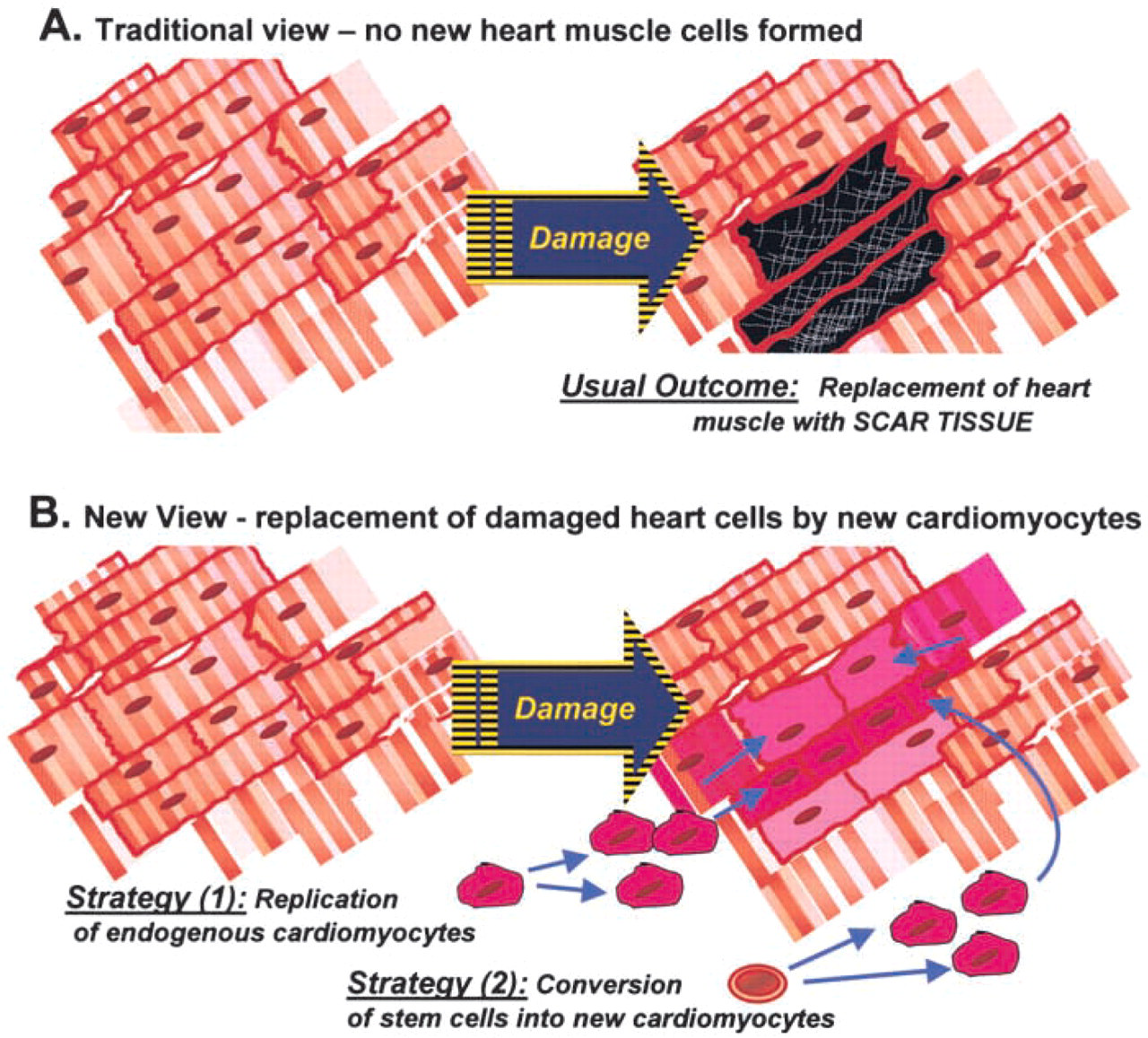
Repair of damaged heart muscle.
Transplantation of Skeletal or Fetal Cardiac Muscle Cells into Hearts
Skeletal Muscle. Attempts to use autologous skeletal muscle to repair damaged hearts has been attempted by relocating (from their normal position) wraps of whole sheets of the latissimus dorsi muscle to supplement myocardial function (Astra and Stephenson 2000; Oakley and Jarvis 2001). This process, called cardiomyoplasty, was pioneered in the 1980s (Sola et al. 1985) and has been applied clinically with varied success.
Alternatively, transplantation of isolated cultured myoblasts has been used to try to restore muscle function in hearts (Koh et al. 1993; Robinson et al. 1996; Taylor et al. 1998; Hutcheson et al. 2000; Jain et al. 2001) both experimentally and in the clinical situation. This requires the myoblasts to be extracted from skeletal muscle, expanded in tissue culture, and then usually injected into the heart muscle (Hutcheson et al. 2000). Delivery via the circulation is an alternative option for delivery of donor myoblasts to the whole heart (Robinson et al. 1996). In all cases, however, one of the critical goals must be electromechanical coupling through gap junction communication between the engrafted skeletal myoblasts and the surrounding cardiac myocytes (Reinecke et al. 2000; Suzuki et al. 2001). Further steps in skeletal muscle transplantation are continuing. Recent studies have improved the survival of transplanted myoblasts by heat shock pretreatment (Suzuki et al. 2000), modified their immunoprotection, and confirmed that the benefits of skeletal myoblast transfer are additive with those of conventional therapies, such as angiotensin-converting enzyme inhibition (Pouzet et al. 2001). Autologous myoblast transplantation has now been reported in one human patient, with promising improvements in cardiac function for 5 months (Menasche et al. 2001).
Cardiomyocytes. Attention has recently shifted from such studies using skeletal muscle to the use of cardiomyocytes, and especially stem cells, as a source of replacement muscle. Because traditionally mature cardiomyocytes are considered to be incapable of cell replication, the donor cardiomyocytes to date have largely been of embryonic/fetal origin. Although some experiments in animal models report successful engraftment and maturation of embryonic cardiomyocytes in normal and injured hearts (Soonpaa et al. 1994; Li et al. 1997b; Matsushita et al. 1999), other studies show that most of the donor cardiomyocytes (engrafted into mature rat hearts after infarction) retained their embryonic phenotype and did not form junctions with mature heart cells by 4 weeks (Etzion et al. 2001). Although neonatal donor cells could form junctions with host myocardium, there was massive initial death of donor cells and at later times the grafts were often isolated by scar tissue (Reinecke et al. 1999; Zhang et al. 2001; commentary by Thurmond et al. 2001). This problem is a direct result of the inflammation and scarring after infarction, and it may be that use of cardiomyocyte transplantation therapy could be more effectively developed to address functional improvement in myopathic heart diseases (Yoo et al. 2000). Alternatively, possible engineering of cardiac grafts within three-dimensional scaffolds or aggregates might provide a superior source of donor cells for repopulation of the infarcted heart (Leor et al. 2000; Watzka et al. 2000).
The use of skeletal muscle cells to repair damaged heart muscle has had some success, and experiments are ongoing. A more attractive approach is the use of donor cardiomyocytes, but mixed results have been obtained with donor fetal/embryonic/neonatal cardiomyocytes and the use of such donor cells in humans raises major ethical issues.
Proliferation of Mature Cardiomyocytes
As outlined above, in contrast to the well-documented capacity for regeneration of (post-mitotic) skeletal muscle from muscle precursor (satellite) cells, it has long been considered that proliferation of cardiac myocytes ceases permanently soon after birth and that there is no replacement with new cardiomyocytes after damage. Although binucleated cardiomyocytes apparently arise by nuclear division, cytokinesis is also required to generate new cardiomyocytes. It now appears that there is a low level of myocyte proliferation (accompanied by cytokinesis) in the postnatal heart (Kajstura et al. 1998; Soonpaa and Field 1998; Beltrami et al. 2001). Observation of cells undergoing mitosis reveals that proliferating cardiomyocytes represented about 14 cells per million in normal heart and that this was increased 10-fold in heart disease. In the left ventricle of a 45-year-old man there are about 5 × 109 myocyte nuclei, and a mitotic index of 14 nuclei/106 cells implies that over 81,000 nuclei are in mitosis at any given time. Because mitosis lasts for about 1 hr, an impressive number of new myocytes may be produced each year. Since there is also a progressive loss of cardiomyocytes, calculated at 6.4 × 106 cardiomyocytes per year (Kajstura et al. 1998), it now appears that there may indeed be a slow turnover of cardiac myocytes throughout life (Soonpaa and Field 1998; Pasumarthi et al. 2000). Although it can be very difficult to distinguish replicating cardiomyocytes from other replicating mononucleated cells in the heart (Soonpaa and Field 1998), detailed studies of postmortem hearts further support increased cardiomyocyte replication in response to heart damage (Beltrami et al. 2001; discussed in Rosenthal 2001). Clinically, this level of cardiomyocyte proliferation seems unable to rescue damaged muscle (Rosenthal 2001). However, the identification of proliferating populations of cardiomyocytes in normal postnatal hearts does raise the possibility of expanding such cells in vivo or of stimulating such cells for cell transplantation to repair heart muscles damaged after ischemia and cardiovascular disease (Reinlib and Field 2000). It is interesting that such replicating cardiomyocytes appear to be increased in transplanted hearts (Quaini et al. 2002).
Many studies have investigated the molecular basis of the “block” to proliferation that occurs in most cardiomyocytes shortly after birth. Increased cardiomyocyte replication is reported after increasing IGF-I levels (Reiss et al. 1996), manipulating cell cycle-regulatory molecules such as the cyclins, associated kinases, and their inhibitors (Poolman and Brooks 1998; Pasumarthi et al. 2000; Liao et al. 2001), and telomerase reverse transcriptase (Oh et al. 2001; reviewed in Schneider and MacLellan 2001), and in injured hearts of adult MRL mice (Leferovich et al. 2001). Furthermore, overexpression of loxP-flanked SV40 T-antigen driven by the cardiac-specific promoter Nkx2.5 caused multiple tumors of the ventricular myocardium at 3–4 weeks postnatally (Markham et al. 2001). Cells isolated from these tumors continued to proliferate in culture until T-antigen expression was removed by Cre recombinase-driven excision. This loss of T-antigen was accompanied by dramatic slowing of proliferation, cell enlargement, and the partial differentiation towards a cardiac myocyte phenotype.
Beyond nuclear replication is the necessity for generation of daughter nuclei (rather than increased ploidy of the original nucleus) and cytokinesis to generate new cardiomyocytes. Therefore, factors that stimulate both of these events need to be investigated. It is not clear whether reversal of the apparently post-mitotic state of cardiomyocytes and cleavage of this highly organized muscle cell may have parallels with the reversal of the post-mitotic status of myonuclei in the sarcoplasm of myotubes/myofibers (as discussed above).
Overall, in contrast to long-standing dogma, there is now evidence to support some proliferation of postnatal mammalian cardiomyocytes in vivo, especially after heart damage. It remains a major challenge to achieve significant proliferation of such postnatal mammalian heart cells. Under the circumstances, an attractive alternative is the use of stem cells to generate new cardiomyocytes.
Cardiomyocytes from Stem Cells
Bone Marrow-Derived Stem Cells. Since the 1999 demonstration of cardiac muscle formation from circulating bone-marrow cells (Bittner et al. 1999) there have been many reports related to stem cell contribution to cardiac muscle formation in rodents and, more recently, in humans. There are few alternative sources of cardiomyocytes (Table 1), in contrast to the broader plasticity seen for skeletal muscle, and such alternative precursor cells are not necessarily stem cells (as already discussed for skeletal muscle).
In experimental infarcted hearts in adult mice, cardiomyocytes and vascular cells can be formed in vivo from circulating mouse bone marrow stem cells (Goodell et al. 2001; Jackson et al. 2001) and these stem cells also give rise to cardiomyocytes after direct injection into damaged heart tissue (Tomita et al. 1999; Orlic et al. 2001; Toma et al. 2002). It appears that cardiomyocytes can be formed from bone marrow-derived HSCs, MSCs, or endothelial stem cells. A key issue is clearly the extent to which this can occur in vivo. Systemic delivery of highly purified HSCs in lethally irradiated mice contributed to the formation of cardiomyocytes and endothelial cells in ischemic hearts (Jackson et al. 2001). In rats, transplantation of rat bone marrow cells directly into cryo-damaged heart improved heart function with increased cardiac myocyte cells and angiogenesis in the scar (Tomita et al. 1999), and related experiments after ischemic heart injury in mice showed many cardiomyocytes and endothelial and vascular smooth muscle cells derived from injected (green fluorescent protein-labeled) donor bone marrow cells (Orlic et al. 2001). Mesenchymal (stromal) stem cells from bone marrow injected directly into rat hearts also gave rise to cardiomyocytes (Wang et al. 2000), and this has also been demonstrated for human bone marrow-derived MSCs (Toma et al. 2002). Therefore, both bone marrow-derived HSCs and MSCs can give rise to cardiomyocytes, although it should be noted that the bone marrow MSCs are probably not normally present in the bloodstream. Transplantation of human bone marrow endothelial stem cells into rat heart formed new vasculature but not cardiomyocytes, and improved cardiac function (Kocher et al. 2001). Because there appears to be a close relationship between endothelial cells and cardiomyocytes (see below), it remains to be clarified whether such bone marrow-derived endothelial stem cells can indeed form cardiomyocytes in some situations. As already discussed (with respect to skeletal muscle), there is a close and complex relationship between bone marrow-derived HSCs and endothelial stem cells (Nishikawa 2001), and also between MSCs and endothelial stem cells (Reyes and Verfaillie 2001).
Although the formation of vascular cells such as endothelial cells may not appear directly relevant to the attempts to repair damaged hearts, it should be noted that an adequate blood and oxygen supply for newly seeded cardiomyocytes is critical for their survival. Therefore, the ability of transplanted bone marrow stem cells to form the cells of the vasculature is another important advantage to the use of a totipotent cell type (Kamihata et al. 2001; Kocher et al. 2001), and in some instances it has been shown that the establishment of new endothelial cells is more common than the formation of new cardiomyocytes (Jackson et al. 2001). There are already reports in the popular press of autologous bone marrow stem cells of a patient being injected directly into his heart during coronary bypass surgery, although details of the outcome/benefits of this are lacking (G. Steinhoff and M. Freund in Germany, reported by Reuters July 23rd, 2001).
Female hearts transplanted into male recipients provide the opportunity to assess the contribution of host male (Y-chromosome-positive) cells to undamaged myocardium of the donor heart in a clinical context. In contrast with earlier studies, Quaini et al. (2002) reported a significant (7–10%) and rapid (by 4 days) contribution of host (male) cells to myocytes and blood vessels in such human female heart transplants. One potential criticism of these observations is the possibility that the female donor hearts may have already contained male cells before transplantation. These male fetal cells can be derived from trophoblasts or nucleated erythrocytes during gestation. They are detected in maternal blood within 6 weeks after conception, a large fetomaternal transfusion occurs at the time of labor and delivery, and microchimerism in the mother is documented for at least 27 years (Bianchi 1998; Nelson 2001). Therefore, it would be of considerable interest to know if the donor females and control females whose hearts were used in the study by Quaini et al. (2002) had indeed experienced a male pregnancy (even if it did not go to term). In addition, careful examination of female hearts (in the absence of transplantation) could reveal the potential contribution of such male cells to cardiac chimaerism in these mothers (Bianchi 1998).
Endothelial Cells. The interesting demonstration of good conversion (trans-differentiation) of mouse and human endothelial cells into cardiomyocytes in tissue co-culture and in vivo (Condorelli et al. 2001) raises the real possibility of using human endothelial cells for cardiac repair. This study emphasized the importance of cell contact between the endothelial cells and existing cardiomyocytes for such conversion. Whereas embryonic and neonatal endothelial cells are capable of forming cardiomyocytes, endothelial cells isolated from mature animals are not, indicating a marked loss of plasticity during development. However, relatively well-differentiated endothelial cells derived from human umbilical veins did form cardiomyocytes (Condorelli et al. 2001). Therefore, a possible source of human endothelial cells for therapeutic purposes is the umbilical cord, and ideally these would be autologous cells obtained from an archival cord bank. Alternatively it might be possible to expand populations of circulating human endothelial progenitor cells (Kawamoto et al. 2001).
Embryonic Stem Cells. It is well established that murine embryonic stem (ES) cells can give rise to cardiomyocytes in vitro and in vivo (Klug et al. 1996; reviewed in Hescheler et al. 1999). It has now been shown in tissue culture that human ES cells can also differentiate into cardiomyocytes (Hescheler and Fleischmann 2001; Kehat et al. 2001). However, human ES cells have a very low efficiency of conversion into cardiomyocytes compared with those of mice. As Kehat and colleagues have noted, there are striking differences in the human and murine stem cell models, and this must be considered in evaluating experiments in mice to develop strategies to improve human heart function. For example, human ES cells differentiate in the absence of a feeder layer and in the presence of leukemic inhibitory factor. Other notable differences include the lower number of human ES cells able to undergo differentiation and spontaneous contraction (<10% compared with >80% of murine ES cells) and the slower time course of differentiation (a median of 11 days compared with 2 days for murine cells). Whether these differences reflect true species differences, or simply our current understanding of these cell systems, or different microenvironment requirements, remains to be determined. The use of embryonic stem cells as a source of cardiomyocytes (and many other cell types) is an attractive therapeutic possibility that needs to be fully explored.
Neural and Hepatocyte Stem Cells. Other studies have shown that a clonal hepatocyte stem cell line WB-F344, when transplanted into the hearts of mice in vivo was able to differentiate into cardiac myocytes (Malouf et al. 2001). Furthermore, although cardiomyocytes can be derived from stem cells of neural tissues during development (Clarke et al. 2000) and in experimental situations, the efficacy is relatively low (Condorelli et al. 2001).
In summary, given the time constraints for repair after acute myocardial infarction, delivery of pre-differentiated cells (cardiomyocyte and vascular cells possibly derived from stem cells) appears desirable. Local delivery of these cells results in direct seeding of the damaged zone, but we need to understand more about how the microenvironment promotes cell differentiation so that this can be exploited. Local delivery might be improved if such cells were engineered into 3D grafts on appropriate matrix/biomaterials.
Systemic delivery of stem cells is relatively non-invasive and remains an attractive option. This relies heavily on the ability of cells to home to damaged tissue. However, little is known yet about the factors responsible for such specific tissue targeting. Furthermore, expansion of the stem cell population in the damaged heart and differentiation into functional car-diomyocytes are required, and the local conditions that dictate this need to be elucidated.
For heart tissue there have been some rather promising results in the past 3 years with bone marrow and endothelial stem cell replacement of cardiomyocytes and vascular cells. These studies need to be firmly substantiated in vivo and carefully evaluated for humans. It is early days and, in the absence of solid in vivo data, it seems premature to extend such studies to the clinical situation.
Footnotes
Acknowledgements
The research on myoblast transfer therapy and stem cells was made possible by grants from the Association Francaise Contre les Myopathies (MDG), the International Duchenne Parent Project (MDG), and the Muscular Dystrophy Association of America (MDG and NR). Recent funding from the Heart Foundation of Australia (MDG, MB, and NR) is also acknowledged.
The generous comments and unpublished data of colleagues are gratefully acknowledged. We are particular grateful to Zipora Yablonka-Reuveni for helpful criticism.