
Abstract
T
AE proteins are encoded by multiple transcripts that appear to be expressed in a tissue- or cell type-specific fashion (Kudrycki et al. 1990; Linn et al. 1992; Cox et al. 1995; Wang et al. 1996). Thus, AE1 (originally termed Band 3) is expressed mainly in erythrocytes and its N-terminally truncated variant in kidney epithelial cells. AE3 is expressed predominantly in neurons and its N-terminally truncated variant in heart. The most ubiquitously expressed AE in mammalian cells is AE2, which is found in almost all cells and tissues studied thus far. In rat, three AE2 splice variants have been identified (Wang et al. 1996; see also Figure 1). These include the AE2a variant, which corresponds to the full-length AE2 protein (1241 aa), AE2b, which has an alternative promoter in intron 2 and therefore lacks the first 17 amino acids of AE2a, and AE2c, which has an alternative promoter in intron 5, resulting in a 1035-aa protein. In human, two AE2b variants [AE2b(1), AE2b(2)], which are closely related to the rat AE2b variant, have been recently identified (Medina et al. 2000). The two variants differ slightly from each other within their N-termini. AE2c variant has not been detected in humans.
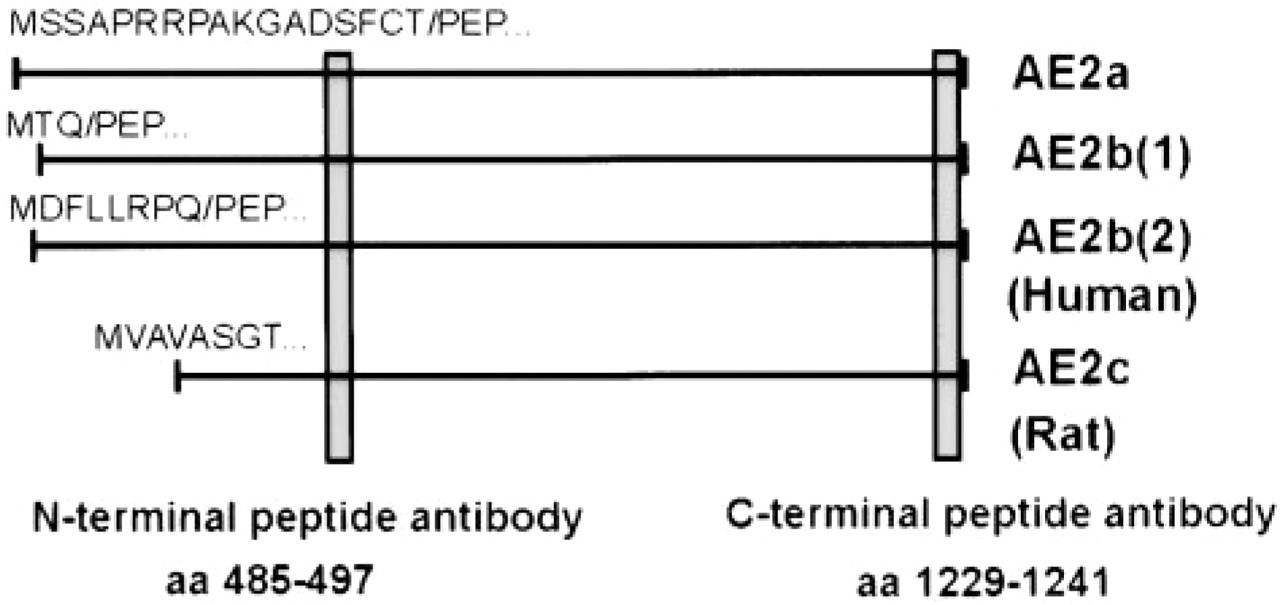
Schematic presentation of the different AE2 variants detected in rat and humans (see text for references). The localization of N- and C-terminal anti-AE2 antibodies used is also shown.
AE polypeptides also show remarkable variability in their subcellular localization in cells and tissues. For example, in kidney tubules, the N-terminally truncated variants of AE1 and of AE2 are localized to the basolateral plasma membrane of polarized epithelial cells (Alper et al. 1999) whereas, in hepatocytes, biliary epithelial cells, and intestinal epithelia AE2 (most probably the AE2a variant), appears to be localized to the apical plasma membrane (Chow et al. 1992; Martinez–Anzo et al. 1993). Previous work from our laboratory has shown that AE2a is also expressed in a highly polarized fashion in the equatorial segment of mammalian sperm cells (Holappa et al. 1999). On the other hand, our previous studies have suggested the existence of an AE1- or AE2-related protein in the Golgi membranes in a range of cell types (Kellokumpu et al. 1988). Recently, similar findings have been reported by others (Alper et al. 1997) using rat kidney as a target tissue. An AE1-related polypeptide has also been detected in mitochondrial membranes (Ostedgaard et al. 1991).
The biological rationale for the existence of multiple AE size variants is not known. One possibility is that the N-terminal sequences that are variable among the transcripts contain regulatory elements (e.g., phosphorylation sites) that can be used either to inhibit or to stimulate their anion transport properties in specified target cells (Green and Kleeman 1992). Alternatively, N-terminal sequences may contain targeting information that is used to localize these protein variants either to the apical or to the basolateral domain in polarized epithelial cells, or to intracellular membranes in others (Cox et al. 1995). Targeting of some AE variants may also involve their direct association with the two well-known cytoskeletal proteins ankyrin and spectrin, whose specific isoforms have recently been identified also in the Golgi and associated transport vesicles (Beck et al. 1994, 1997; Devarajan et al. 1996), since AE polypeptides are generally known to interact with these cytoskeletal proteins via their N-terminal sequences (Bennett 1992; Jons and Drenckhahn 1998). To resolve between the above possibilities, detailed information about the subcellular distribution of each of the known AE size variants is necessary. In this study we set out to identify the Golgi-associated AE in human fibroblasts and found that it is the full-length AE2a isoform. These results provide compelling evidence for the existence of an AE in the Golgi membranes.
Materials and Methods
Antibodies and Reagents
The antibody against the AE2 C-terminal peptide (amino acids 1229–1241 of AE2) was prepared as described (Parkkila et al. 1993). Briefly, the peptide was coupled to ovalbumin, emulsified with Freund's complete adjuvant, and injected intradermally into rabbits. Four booster injections in incomplete adjuvant were given at 3-week intervals. The antibody was affinity-purified with the C-terminal peptide coupled to Affigel-10 beads (BioRad; Hercules, CA) and eluted with 4 M MgCl2. The eluate was desalted immediately using a Sephadex G-25 column.
An N-terminal peptide antibody against AE2 (amino acids 485–497 of AE2) was a kind gift from William Horne (Yale University; New Haven, CT). C-terminal and N-terminal AE1-specific polyclonal and monoclonal antibodies were generous gifts from Drs. Ron Kopito (Stanford University; San Francisco, CA), Marcin Czerwinski (BIII.136; Ludwik Hirszfeld Institute of Immunology, Wroclaw, Poland), and David Anstee (Bric 130, Bric 169; International Blood Group Laboratory, Bristol, UK).
A monoclonal anti-GFP antibody and the EGFP-C1-vector were obtained from Clontech. Tetramethylrhodamine isothiocyanate (TRITC)-conjugated swine anti-rabbit and fluorescein isothiocyanate (FITC)-conjuaged goat anti-mouse secondary antibodies were obtained from Dakopatts (Copenhagen, Denmark). Peroxidase-conjugated sheep anti-rabbit and goat anti-mouse antibodies were from BioSys (Compiegne, France). Antibodies against β-COP (Affinity Bioreagents; Denver, CO) and giantin (a kind gift from Dr. Hans-Peter Hauri; University of Basel, Switzerland) were used as Golgi markers.
Stock solutions of cycloheximide, nocodazole (Sigma, St Louis, MO; both 10 mg/ml) and brefeldin A (Boehringer–Mannheim, Mannheim, Germany; 5 mg/ml) were dissolved in ethanol, stored at –20C, and used at 1:1000 dilution.
Cell Cultures
Human embryonic skin fibroblasts were grown in MEM (Gibco; Grand Island, NY) with glutamax supplemented with 10% newborn calf serum, penicillin-streptomycin (Gibco), and 50 μg/ml ascorbic acid. Treatment of cells with cyclo-heximide, brefeldin A, and nocodazole was for 1–5 hr, 30 min, and 4 hr, respectively. The COS-7, NRK, and CHO-K1 cells were obtained from ATCC (Rockville, MD) and were cultured in media recommended by ATCC.
Indirect Immunofluorescence
Cells were processed for indirect immunofluorescence as described elsewhere (Kellokumpu et al. 1988). All incubations, unless otherwise stated, were carried out in the presence of 0.05% saponin. Stained specimens were examined using an epifluorescence microscope (Leitz Aristoplan; Wetzlar, Germany) equipped with appropriate filters or with a Leitz CLSM confocal microscope (Leica Laser Technics; Heidelberg, Germany).
Northern Blotting Experiments
Isolation of total RNA from cells and human testicular tissue (obtained from Oulu University Hospital) was performed using the acid guanidinium thiocyanate–phenol–chloroform extraction method. Northern blotting experiments were performed as previously described (Parkkila et al. 1993). Briefly, 20 μg of total RNA from each sample was size-separated by electrophoresis and transferred onto a nitrocellulose filter in high salt buffer (10 × SSC). After baking, the filter was prehybridized and hybridized at 42C with [32P]-CTP-labeled AE2 cDNA probe (Random Priming Kit; Boehringer-Mannheim). The probe corresponds to a portion of the membrane-spanning domain of AE2 (nucleotides 3080–3509 of the AE2a cDNA). Hybridization buffer consisted of 50% formamide–5 × SET-5 × Denhardt's –0.1% SDS–50 μg/ml tRNA. Washings were done at 42C in 2 × SSC for 3–4 hr.
RT-PCR
RT-PCR was utilized to amplify overlapping fragments of AE2 cDNA (each about 1 kb in length) from total RNA samples isolated either from human testicular tissue or from cultured human fibroblasts. For reverse transcription, 1 μg of total RNA was used, and two subsequent PCR rounds were performed with AE2-specific primer pairs. For the second round PCR, one of the primers in each primer pair was nested with respect to the first-round primer. As a control template, we used the full-length human testicular AE2a cDNA inserted in pGEM-4Z plasmid vector (Holappa et al. 1999). The cycling parameters used were 1 min at 94C, 1 min at 58C, and 1 min at 72C for the first round PCR and 1 min at 94C, 1 min at 65C, and 1 min at 72C for the second-round PCR. The following second-round primers were used: primer F13 (nucleotides –47 to +26); 5′-GTTGCCCTGAATTCCGCAGCGA-3′, R12 (nt 907–882); 5′-CTTCTCGGCCACTCTGTGTGGAACCT-3′, F23 (nt 691–711); 5′-CGCAGCTACAACCTTCAGGAG-3′, R45 (nt 1808–1789); 5′-TTGATGGCCGTCAGCAGGTC-3′, F47 (nt 1686–1706); 5′-GAGTAGTGCCAACATGGACTA-3′, R31 (nt 2456–2436); 5′-ACGAAGCGGACCAGGAAGCTC-3′, F42 (nt 2379–2399); 5′-TGTGTGGATCGGCTTCTGGCT-3′, R43 (nt 3590–3573); 5′-ACTGTGAGGATGAGGATG-3′ (F = forward primers; R = reverse primers). Numbers denote the base numbering of the human full-length AE2a cDNA. The exact sizes of the expected PCR products were calculated to be 954 bp, 1118 bp, 771 bp, and 1212 bp, respectively. Together, the amplified sequences cover almost the entire coding region (starting 47 nucleotides before the translational initiation codon) except the last 136 nucleotides just before the stop codon.
Antisense Oligonucleotide Treatments
Phosphorothioate antisense oligonucleotides (24-mer; 5′-GCGCTGCTCATGGCCGAATCTTAG-3′) directed against the translational initiation site and flanking sequences of the AE2a mRNA were ordered from Amersham-Pharmacia Biotech (Poole, UK) and manufactured by Genosys Biotechnologies. A nonsense oligonucleotide (22-mer) was used as a control in the experiments. Both antisense and nonsense oligonucleotides (25 μg/ml) were added directly to 1-day-old COS-7 cell cultures and the cells were then cultivated in medium containing serum for 3 additional days before washing with PBS, fixation, and analyses by indirect immunofluorescence with the anti-AE2 antibody.
GFP Fusion Protein Constructs and Transfections
To prepare the full-length AE2-GFP fusion protein, pGEM-4Z plasmid containing human AE2a cDNA (cloned from human testicular tissue) was first mutated by using the Quick Change mutagenesis kit (Stratagene; La Jolla, CA) to eliminate the stop codon just before the ATG translational initiation site and to create a new 5′-HindIII site for insertion into the GFP vector (pEGFP-C1; Clontech, Palo Alto, CA). The mutated full-length AE2 cDNA was subcloned into the EGFP-C1 vector after digestion with EcoRI and HindIII restriction enzymes. After transformation into JM 109 cells, cDNA was isolated using the plasmid purification kit of Qiagen. Transfections into COS-7 cells plated 1 day earlier were done using 1–2 μg of purified AE2a cDNA/30-mm plate and the FUGENE6-transfection reagent (Boehringer–Mannheim) according to the manufacturer's instructions. Transfected cells were examined 20–24 hr after transfection.
Immunoblotting
Cells were washed and scraped directly into hot SDS sample buffer with a rubber policeman. The sample was collected, vortexed rigorously for 2 hr to eliminate viscosity, and heated for 3 min at 94C before subjection to SDS-PAGE (7.5–9% acrylamide gel). Size-separated samples were transferred onto a nitrocellulose filter and immunoblotted with the anti-GFP and anti-AE2 antibodies. Stained bands on the blots were visualized using the ECL detection system (Amersham) and FUJI RX X-ray film.
Trypsin Treatments and Cell Surface Biotinylation
To quantitate the level of expression of the fusion protein at the cell surface, transfected COS-7 cells were treated on plates at either 4C or 37C with 0.2% trypsin for 30–60 min. Cells were then collected by centrifugation, rinsed with PBS, and solubilized directly into SDS sample buffer and analyzed by SDS-PAGE and immunoblotting with the anti-GFP antibody. As an alternative approach, transfected cells were biotinylated using 0.5 mg/ml sulfo-NHS-biotin (Pierce; Rock-ford, IL) in PBS on ice. Cells were dissolved in 0.5% Triton X-100-0.5% deoxycholate in PBS and biotinylated proteins were absorbed by using immobilized streptavidin (Boehringer–Mannheim). Unbound and absorbed material was collected and analyzed by SDS-PAGE.
Detergent Extraction with Triton X-100
Fibroblasts and COS-7 cells were subjected to detergent extraction protocol (Beck et al. 1994) as follows: 35-mm plates at about 50–70% confluency were first washed three times with PBS at 20C and cooled on ice for 10 min. One ml of ice-cold 1% Triton X-100 (in 20 mM Tris–10 mM KCl–1 mM MgCl2, pH 7.4) was then added to each plate and incubated for an additional 30 min in a rotator. In some experiments, 0.3 M and 1 M KCl was included in the above buffer. Detergent solutions were collected and the remaining cells on plates were then fixed for indirect immunofluorescence with the anti-AE2 antibodies. Control cells were fixed before the extraction protocol. To quantitate the amount of AE2a protein that could be solubilized with Triton X-100, we used transfected COS-7 cells and extracted cells sequentially with Triton X-100 and SDS sample buffer. Equivalent amounts of both fractions were then used for immunoblotting analyses with the anti-GFP antibody. The intensity of GFP signal in both fractions was quantitated by using an image analysis program (MCID; St. Catharines, Ontario, Canada).
Results
Both N- and C-terminal Antibodies Against AE2 Stain the Golgi Membranes in Different Cell Lines
Previously, the presence of a Golgi-associated AE was detected by using a C-terminal peptide antibody against the erythrocyte AE1 (Kellokumpu et al. 1988). In this study, we obtained a similar staining pattern by using an affinity-purified anti-AE2 C-terminal antibody raised against a synthetic AE2 C-terminal peptide. This antibody (anti-AE2) stained the Golgi complex in many cell types, including cultured human skin fibroblasts (Figure 2A), rat osteosarcoma cells (Figure 2C), and NRK cells (Figure 2D).
Because both of these antibodies (anti-AE1 and -AE2) recognize AE1- and AE2-derived C-terminal peptides (nine of 12 amino acids are identical), it was not possible to ascertain which of the two isoforms (AE1 or AE2) is expressed in the Golgi. Therefore, we next used antisera that were specific for the N-terminal cytoplasmic domains of either AE1 or AE2. However, none of the three N-terminal anti-AE1 antibodies we obtained (see Materials and Methods) stained the Golgi membranes or the plasma membrane in fibroblasts, nor did they recognize any bands in immunoblotting experiments with fibroblast total-cell lysates (data not shown). However, they recognized the human erythrocyte AE1 polypeptide (a 100-kD band). In contrast, an AE2-specific N-terminal peptide antiserum (raised against amino acids 482–497 of AE2) gave Golgi staining in fibroblasts (Figure 2B) and in COS-7 cells (data not shown). These data indicate that the AE2 isoform is the endogenous Golgi-associated AE polypeptide in fibroblasts and in COS-7 cells.
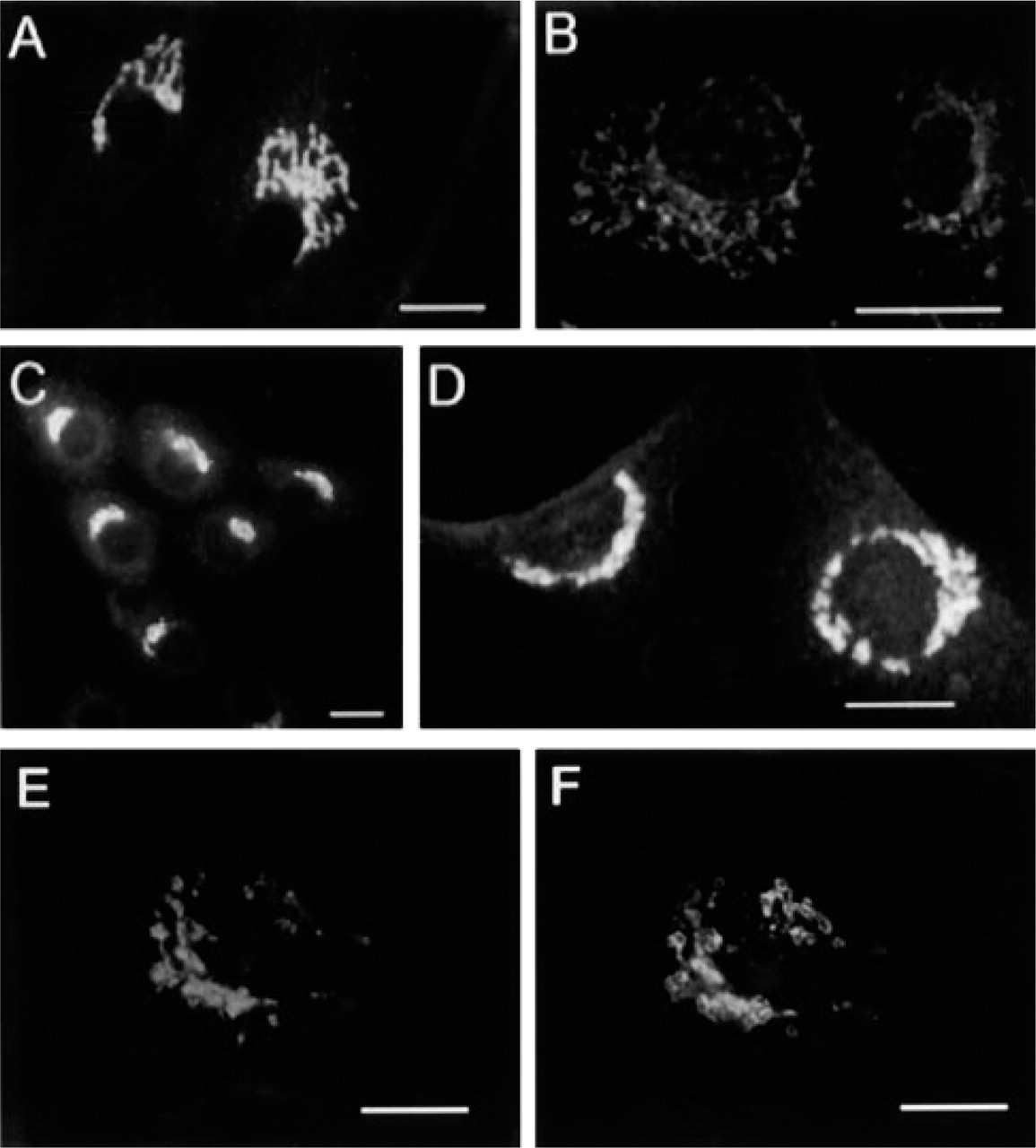
Indirect immunofluorescence staining of cultured cells with the anti-AE2 C-terminal (
Double-staining experiments in COS-7 cells with the anti-AE2 antibody (Figure 2E) and monoclonal antibodies against giantin (Figure 2F), β-COP, and KDEL receptor confirmed that the endogenous AE2 protein also co-localizes with these Golgi markers. Brefeldin A treatment of fibroblasts also caused a rapid redistribution of the AE2 into the ER (Figures 3A–3A). These findings are fully compatible with our previous immunoelectron microscopic data, which have shown that the AE-related antigen was concentrated in the cisternal membranes of the Golgi stack (Kellokumpu et al. 1988).
Finally, we assessed whether the Golgi AE2 is merely in transit towards the plasma membrane by using cycloheximide, a drug that inhibits protein synthesis but does not block transport to the cell surface. Indirect immunofluorescence experiments showed (Figures 4A–4A) that treatment of cells for up to 5 hr did not diminish staining of the Golgi. The concentration of the drug used (10 μg/ml) was found to inhibit incorporation of [35S]-methionine into TCA-precipitable proteins by more than 95%. Therefore, the AE2 protein can be considered a resident of the Golgi membranes.
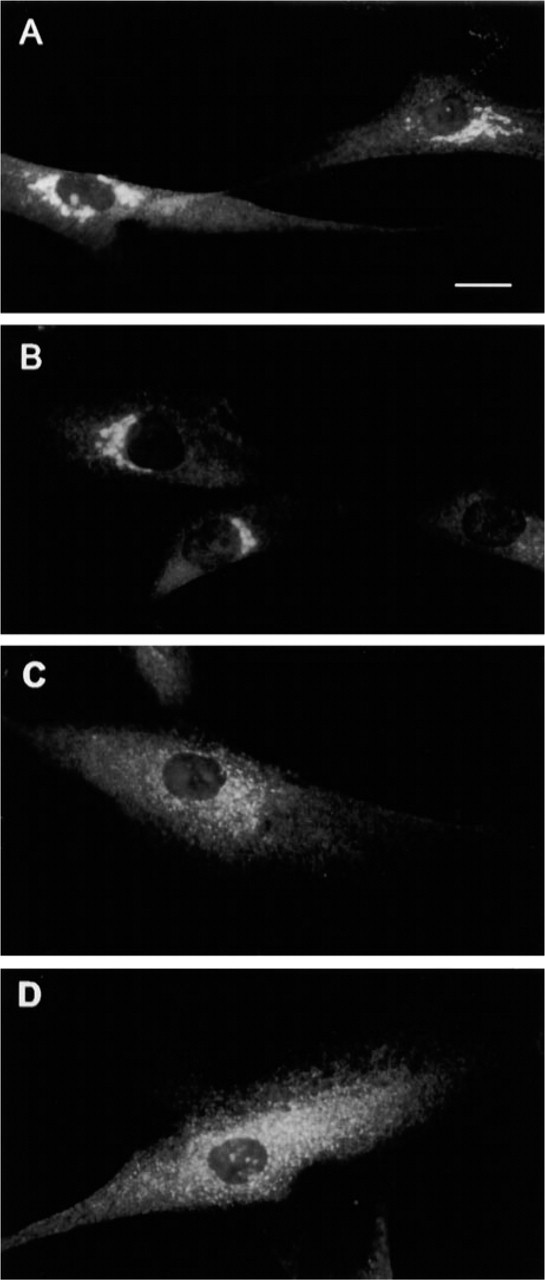
Redistribution of the AE2 protein into the ER in brefeldin A-treated human fibroblasts. Cells were incubated in the presence of BFA (5 μg/ml) for 0 min (
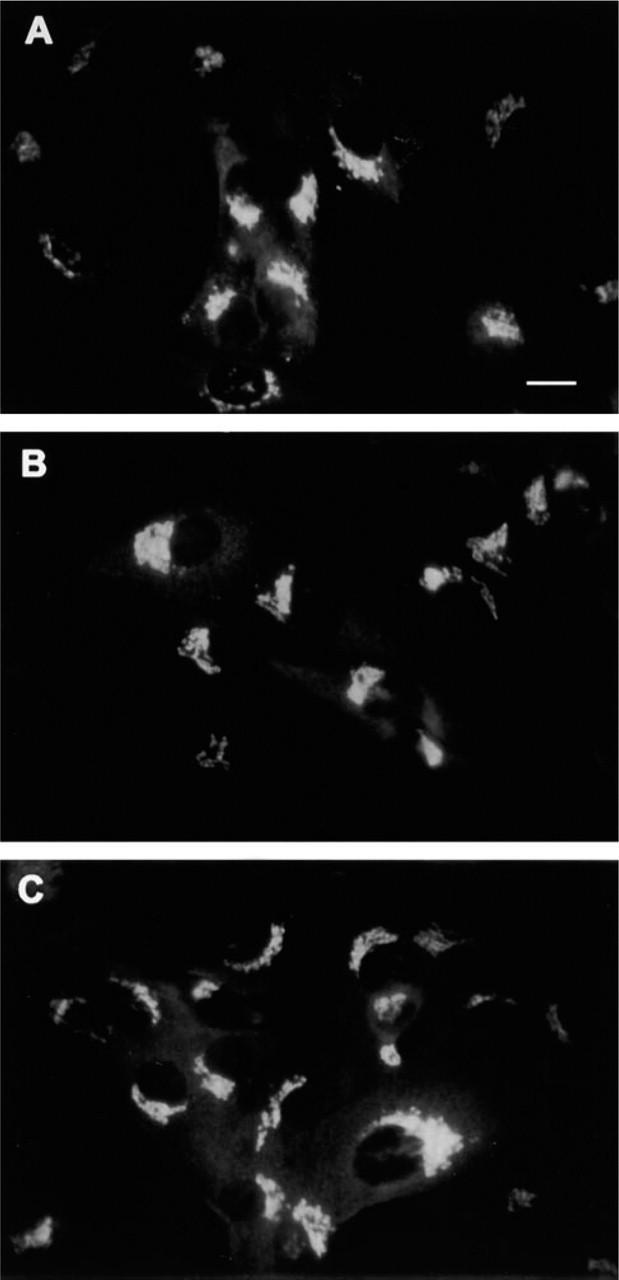
Distribution of the AE2 protein in cycloheximide-treated cells. ROS 17/28 cells were treated with cycloheximide for 0 hr (
AE2a mRNA Is the Predominant mRNA Species in Fibroblasts
Because both the N- and C-terminal antibodies recognize all known AE2 variants (Figure 1), Northern blotting and RT-PCR analyses were necessary to specify more exactly the Golgi-resident AE2 isoform or variant. In three different cell lines that show prominent Golgi staining with antibodies to AE2 (two of which are shown in Figure 2), Northern blotting analyses detected only a 4.4-kb mRNA species with an AE2 cDNA probe that corresponded to a portion of the C-terminal membrane-spanning domain of AE2 (Figure 5A). On the basis of its size, this mRNA species corresponds to the full-length AE2a mRNA transcript. Transcripts of the N-terminally truncated AE2 isoforms in rat (AE2b, 4.2 kb and AE2c, 3.8 kb) and human tissues [AE2b(1) and AE2b(2)] were not detected in these cell lines. It should be noted that the probe used should hybridize with all N-terminally truncated AE2 variants if they were present in these cells.
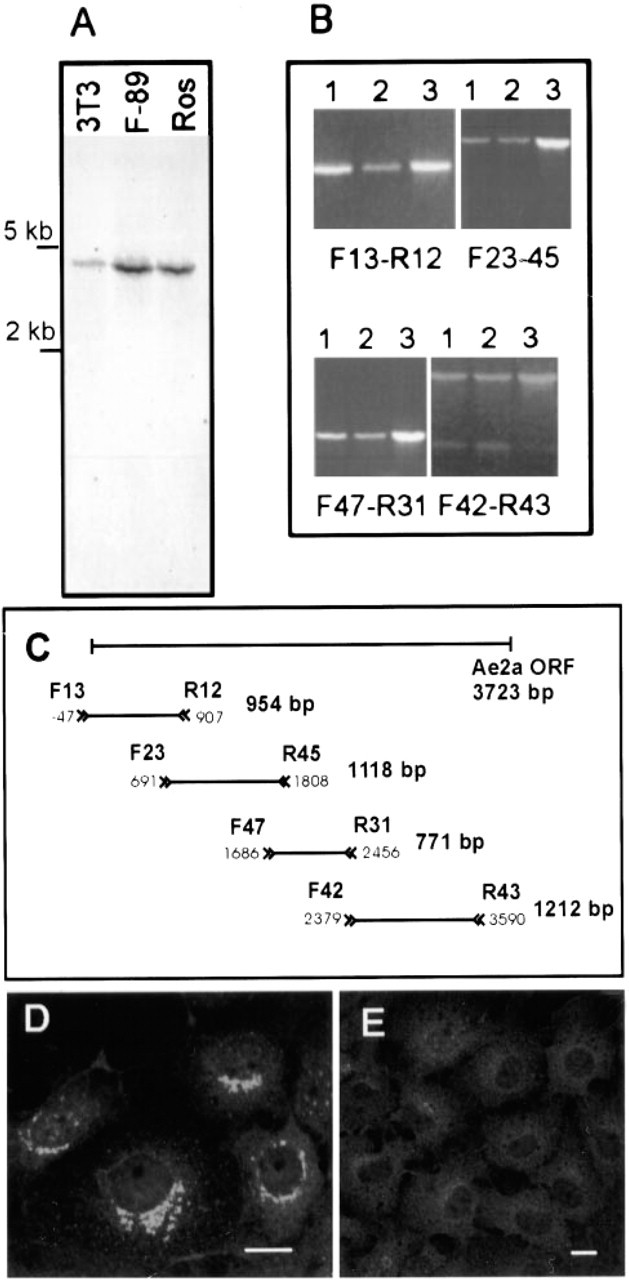
(
To confirm the identity of the 4.4-kb transcript detected as the full-length AE2a mRNA, we next performed RT-PCR to amplify the complete coding region of the AE2a mRNA as four overlapping fragments. For the RT-PCR, we used total RNA isolated either from cultured human fibroblasts or from human testis tissue. Rat testis is known to express mainly the AE2a mRNA (Wang et al. 1996). As a size reference, we used AE2a cDNA, which we have cloned from human testicular lambda gt11 library (Holappa et al. 1999). RT-PCR amplification (Figure 5B) showed that PCR products of identical size were obtained from all three templates, even with the primer that hybridizes only with the 5
Antisense Oligonucleotides Inhibit the Expression of Endogenous AE2 Protein in the Golgi Membranes
To provide additional evidence for the identity of the Golgi-associated AE as the full-length AE2a isoform, we treated COS-7 cells with AE2a-specific antisense oligonucleotides (25 μg/ml) that corresponded to the translational initiation site and flanking regions of the AE2a mRNA. Treatment for 3 days under normal cell culture conditions was sufficient to cause a marked inhibition (≥80%, based on fluorescent intensity measurements) of the expression of endogenous AE2 polypeptide in the Golgi in COS-7 cells (Figure 5E), as evidenced by indirect immunofluorescence staining using the C-terminal anti-AE2 antibody. The effect was specific because nonsense oligonucleotides did not decrease the expression of the Golgi-localized AE2 protein (Figure 5D). Unfortunately, we have been unable to quantitate the inhibition by immunoblotting, most probably because the C-terminal epitope has been reported to be sensitive to SDS (Alper et al. 1997).
Transient Expression of GFP-tagged AE2a Fusion Protein
Indirect immunofluorescence with the anti-AE2 antibodies showed consistent staining of the Golgi membranes in COS-7 cells (Figure 5D). These cells therefore express an endogenous AE2 protein in the Golgi membranes. To enable detection and localization of transiently expressed human AE2a protein in these monkey cells, we inserted a cDNA encoding the green fluorescent protein (GFP) into the N-terminus of the human AE2a cDNA cloned from the testicular tissue. This does not affect membrane insertion of the fusion protein because AE polypeptides are known to have an internal signal sequence.
Transient expression of the GFP-tagged full-length human AE2a protein in COS-7 cells showed (Figure 6A) that the fusion protein accumulated in juxtanuclear structures that most probably represented the Golgi membranes. A similar expression pattern was also seen in transfected CHO-K1 cells (data not shown). Accumulation of the GFP–AE2a fusion protein predominantly in the Golgi membranes was confirmed by using both nocodazole and co-localization experiments with antibodies to known Golgi-markers. First, we treated transfected cells for the last 4 hr during the 20-hr transfection with nocodazole, a drug that severs microtubule polymerization. In drug-treated cells, the AE2a fusion protein was found in small punctate structures that were scattered throughout the cytoplasm (Figure 6B). Staining of the transfected cells with antibodies to either giantin (Figure 6D) or β-COP (Figure 6G) also showed that the GFP–AE2a fusion protein co-localizes with the Golgi markers used. However, non-overlapping areas were also detected in transfected cells (Figures 6E and 6H).
Detection of GFP-tagged AE2a Fusion Protein by Immunoblotting
To show that the transfected cDNA encodes a full-size AE2a protein, we next performed immunoblotting experiments with the total cell lysates from transfected COS-7 cells (Figure 7A) by using the anti-GFP monoclonal antibody (Figure 7, Lane 3) and the C- and N-terminal anti-AE2 antibodies (Figure 7, Lanes 2 and 4). All antibodies recognized one major protein of about 200 kD. In untransfected cells, this band was not seen (Figure 7, Lane 1). The size of this band corresponds well with the predicted molecular size of the fully glycosylated AE2a protein (165 kD), given that GFP itself has a molecular size of about 27 kD. The identity of an additional 165-kD band that was detected variably only with the anti-GFP monoclonal antibody (Figure 7A, Lane 3) is not known. The calculated molecular weight of the AE2a protein based on its amino acid composition is 137 kD, so the 165-kD band may represent an unglycosylated form of the GFP-tagged AE2 polypeptide (27 kD +137 kD) or its proteolytic product. It is not precisely known if and to what extent the AE2 protein is glycosylated, but it has four potential N-glycosylation sites, three of which reside in its extensive third extracellular loop.
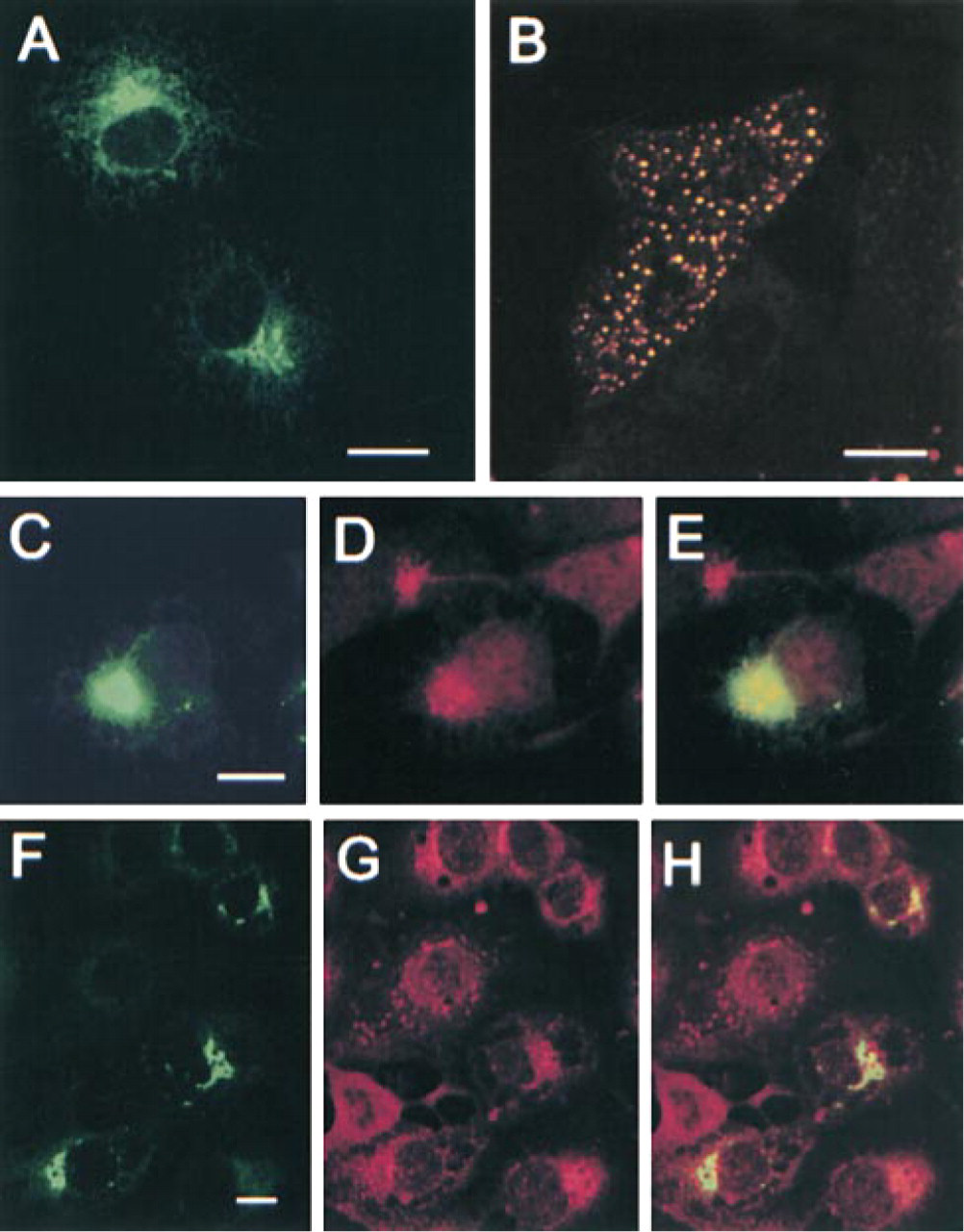
Localization of the GFP-tagged AE2a fusion protein in COS-7 cells. (
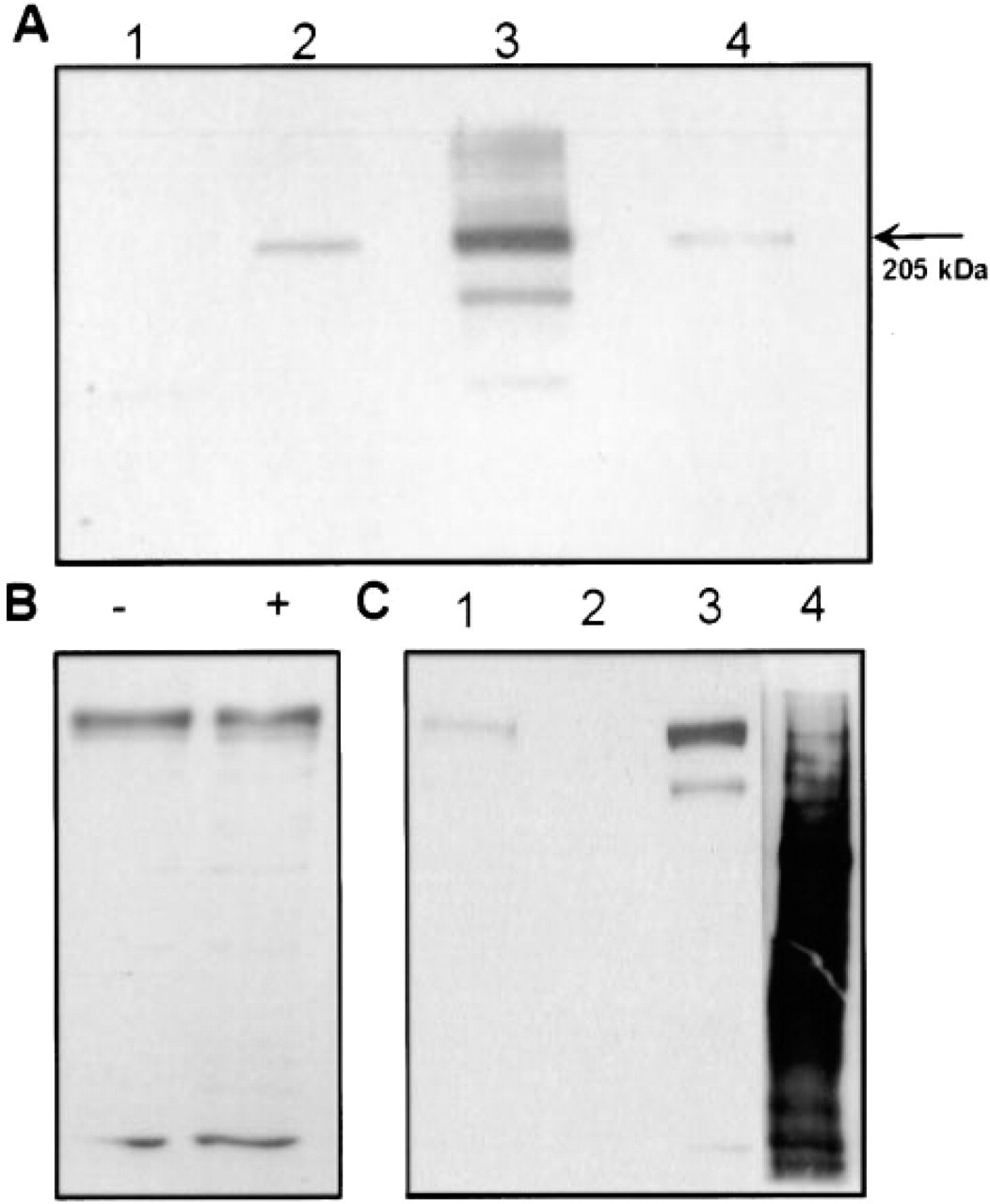
SDS-PAGE and immunoblot analyses of the expressed fusion protein in COS-7 cells. After transfections, cells were lysed directly into Laemmli's SDS sample buffer, and subjected to SDS-PAGE. After transfer to a nitrocellulose filter, the samples were immunoblotted with the anti-AE2 C-terminal antibody (Lane 2), the monoclonal anti-GFP antibody (Lane 3), and with the anti-AE2 N-terminal antiserum (Lane 4). Note that all antibodies recognized a major band with a molecular size of about 200 kD in the transfected cells. Lane 1 shows the control, in which untransfected cells were used as a starting material and immunoblotting was performed with the monoclonal anti-GFP antibody. (
Quantitation of the Cell Surface Expression of the GFP-AE2 Fusion Protein
Because all AE polypeptides are usually considered to be plasma membrane proteins, it was necessary to quantitate the amount of the GFP–AE2a fusion protein that was transported to the plasma membrane in transfected COS cells. Cells transfected with the GFP-tagged AE2a cDNA were subjected on plates to extensive enzymatic digestion with trypsin at 4C or at 37C for 30–60 min. At 37C, this resulted in detachment of the cells from the plates. Subsequent analyses by SDS-PAGE, immunoblotting, and quantitation of the 200-kD fusion protein (Figure 7B) and its potential degradation products showed that it was not degraded by this extracellularly added enzyme. Nevertheless, AE2 contains a number of putative cleavage sites for trypsin, e.g., in its third extracellular domain. On the other hand, permeabilization of the cells with saponin during trypsin digestion resulted in complete degradation of the fusion protein (data not shown). As an alternative approach, we used biotinylation of the cell surface proteins on plates with sulfo–NHS–biotin. We then affinity-purified biotinylated proteins with immobilized streptavidin before SDS-PAGE. Immunoblotting with the anti-GFP antibody showed that the GFP–AE2a fusion protein was not accessible to biotinylation in transfected cells (Figure 7C, Lanes 1–3). In contrast, a number of other cell proteins bound to streptavidin beads and could be detected on the same blots with peroxidase-conjugated streptavidin (Figure 7, Lane 4). These findings indicate that only negligible amounts of the AE2a fusion protein are transported to the cell surface in transfected COS-7 cells.
Detergent Extraction of the Golgi-associated Anion Exchanger
To evaluate whether the Golgi-localized AE2a might be associated with the recently identified Golgi membrane skeleton, we utilized the nonionic detergent extraction protocol (Triton X-100 at 4C) and the poor solubilization properties of many cytoskeleton-associated proteins (Beck et al. 1994). When either fibroblasts (Figures 8A and 8B) or transfected COS-7 cells (Figures 8C and 8D) were solubilized for 30 min on ice with 1% Triton X-100, we found that a substantial portion of the Golgi-associated AE2a polypeptide resisted Triton X-100 extraction and could be detected intracellularly. Similarly, immunoblotting data obtained with transfected COS-7 cells after sequential solubilization with Triton X-100 and SDS showed that about 35–40% of the expressed GFP–AE2a fusion protein could be solubilized from the cells by using Triton X-100 as the detergent (Figure 8E). Addition of salt (0.3–1 M KCl) to the extraction buffer to disrupt putative cytoskeletal interactions resulted in complete solubilization of the fusion protein from the cells (data not shown), suggesting that cytoskeletal proteins may indeed interact with the AE in the Golgi membranes.
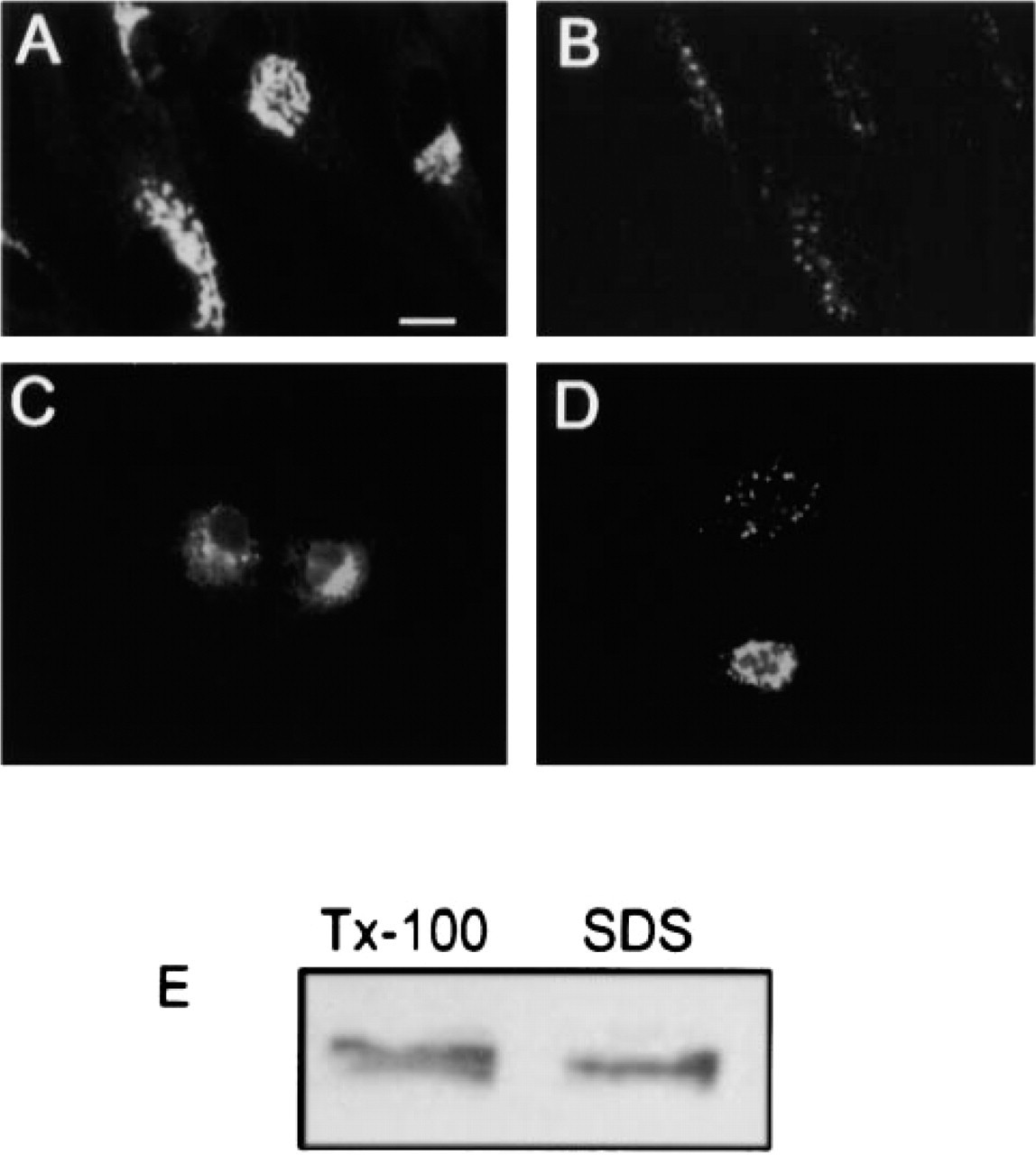
Detergent solubility of the Golgi-localized AE2a. Cultured human skin fibroblasts were extracted on ice with Triton X-100 as described in Materials and Methods. After fixation, cells were stained with the C-terminal anti-AE2 antibodies. Note the strong Golgi staining in untreated cells (
Discussion
In this study, we have identified the full-length AE2a isoform as the Golgi-associated anion exchanger in fibroblasts. The evidence supporting this conclusion is several-fold and includes immunological data (N- and C-terminal anti-AE2 antibodies), mRNA analyses, the use of AE2a-specific antisense oligonucleotides to inhibit endogenous AE2 expression in the Golgi and, finally, the demonstration that the transiently expressed GFP-tagged AE2a fusion protein also accumulated in the Golgi membranes in transfected cells. These data are compatible with our earlier immunological data obtained with the C-terminal anti-AE1 antiserum and with our preliminary sequencing data in rat osteosarcoma cells (Kellokumpu et al. 1988, 1995). The only discrepancy relates to the size of the Golgi-associated AE2 polypeptide identified here (AE2a is a 165-kD protein; Figure 7) and in our earlier work on rat osteosarcoma cells (115 kD). The reason for this is not known, but it may result from the different cell types or antibodies used in these studies (anti-AE1 serum and affinity-purified anti-AE2 antibodies, respectively), or from proteolytic events. Indeed, AE2a appears to be very sensitive to proteolytic attack (Alper et al. 1999). In our hands, successful detection of the AE2a isoform by immunoblotting with the antibodies used (Figure 7) also requires its overexpression in transfected cells.
Overexpression of proteins may lead to formation of aggresomes around the microtubule organizing center (MTOC) if the protein degradation capacity of cytosolic proteasomes is exceeded (Johnston et al. 1998). Protein aggregates formed are delivered to this ubiquitin-rich structure, where they become ensheathed by vimentin. Although the GFP–AE2 fusion protein is also localized in the Golgi region around the MTOC, several facts are against the view that the GFP fusion protein accumulates in aggresomes. First, the endogenous AE2 polypeptide was also found to be located in the Golgi in several cell lines. Second, nocodazole treatment caused dispersal of the GFP-containing structures in transfected cells, although aggresomes are known to be resistant to such treatment. Third, on the basis of its size (200 kD), the expressed GFP–AE2 fusion protein is glycosylated, unlike aggresomal proteins. Finally, ubiquitin did not concentrate in GFP–AE2 fusion protein-containing structures in transfected cells (unpublished observations).
Identification of the AE2a isoform as the Golgiassociated AE raises important questions concerning both its functioning and the apparently variable positioning of different AE polypeptides either in the basolateral or apical plasma membrane in some epithelial cells and, on the other hand, in the Golgi membranes, e.g., in fibroblasts. The vast number of transcripts known to encode AE polypeptides in rat chicken and human tissues (Cox et al. 1995; Wang et al. 1996; Medina et al. 2000) suggests that subcellular localization may depend on the variant in question. Studies on chicken AE proteins have indicated that of the multiple AE1 variants (Cox et al. 1995), only one containing extra exons (exons 5 and 6 encoding part of the N-terminal cytoplasmic domain of chicken AE1) was efficiently retained in an intracellular compartment, probably representing the Golgi. Other variants that lack these exons were expressed at the cell surface. However, the sequence similarity of exons 5 and 6 to that of rat or human AE2a N-terminal sequence is only marginal. Similarly, the AE2c (3.8 kb) variant is the predominant transcript in stomach (Wang et al. 1996), suggesting that it may be the basolateral AE polypeptide in acid-secreting parietal cells.
Alternatively, localization may depend on the cell type in which AE polypeptides are expressed. Studies with kidney AE1 have indicated that the same polypeptide can be sorted either basolaterally or apically and that sorting in this case is guided by external cues (Van Adelsberg et al. 1993, 1994). These findings exemplify the plasticity of protein-sorting machinery in mammalian cells and suggest that positional targeting may also be a cell type-dependent phenomenon.
The positioning of the AE2a isoform in the Golgi membranes in fibroblasts apparently involves specific sorting signals that may cause its oligomerization and oligomerization-dependent retention in the Golgi membranes (Machamer 1991). Similar to many Golgispecific glycosyltransferases, these signals may be buried in any of its multiple transmembrane domains (10–14 spanning regions, some of which appear to consist of less than 20 amino acids) and flanking sequences. Alternatively, oligomerization may be mediated by a leucine-rich region (aa 372–394) within the cytoplasmic N-terminus of the AE2a polypeptide. On the other hand, our detergent extraction data (Figure 8) suggest that AE2a may also associate with cytoskeletal proteins. In this respect, it is intriguing that the cytoplasmic N-terminal domain of AE2a has been shown to bind to erythrocyte ankyrin in blot-overlay experiments (Jons and Drenckhahn 1998), and that Golgi-specific ankyrin (ANK119, ANK195) and spectrin isoforms (erythrocyte β-spectrin, β-III spectrin) (Beck et al. 1994, 1997; Devarajan et al. 1996; Stankewich et al. 1998) have been identified quite recently. The putative interaction of these proteins, as suggested by our detergent/salt extraction data, may therefore be involved in the targeting of the AE2a anion exchanger in the Golgi. In addition, these interactions may be involved in the structural and functional maintenance of the Golgi stacks, in much the same way as their erythroid isoforms control the mechanical stability and cell shape in erythrocytes (Peters et al. 1996).
Footnotes
Acknowledgments
Supported by a grant from the University of Oulu.
We wish to thank Drs R. Baron and W. Horne, D. Anstee, and M. Czerwinsky for kindly providing antibodies used in this study.