
Abstract
P
A cleaved form of uPAR, uPAR(II+III), lacking the amino terminal domain, has been identified in various cell lines and tumors (Høyer-Hansen et al. 1992). Cleavage of uPAR between Domains I and II has been shown to be uPA-dependent in three different cell lines (Høyer-Hansen et al. 1992; Solberg et al. 1994). The functional role of uPAR cleavage is still not known, but recent data indicate that it may be part of an intracellular signaling mechanism influencing cell migration (Resnati et al. 1996; Fazioli et al. 1997).
uPAR has been found on a variety of cell types in vivo, including monocytes and macrophages, granulocytes, keratinocytes, trophoblasts, myofibroblasts (Ploug et al; 1992, Pyke et al. 1994; Rømer et al. 1994; Pierleoni et al. 1998; Dubuisson et al. 2000), and many different tumor cell lines (for review see Ellis et al. 1996). uPA-mediated plasmin proteolysis focused on the cell surface is believed to be involved in a variety of normal and pathological processes that require cell migration and tissue remodeling. Many mouse model systems have been developed to study the function of uPAR in, e.g., tumor invasion and metastasis. However, despite the growing body of information about the biochemical characteristics of uPAR, little is known about expression of the protein under normal physiological conditions in vivo. To address this problem, we have developed a chemical crosslinking assay and generated specific polyclonal antibodies for immunohistochemical localization of uPAR in the mouse.
Materials and Methods
Animals and Tissues
Female BALB/c mice or uPAR gene-targeted mice of mixed 129/Black Swiss background, 6–8 weeks old, were anesthetized with 0.03 ml/10 g of a 1:1 mixture of Dormicum (midazolam 5 mg/ml) and Hypnorm (fluanison 5 mg/ml and fentanyl 0.1 mg/ml) and perfused with cold PBS. Organs for lysate preparations were immediately snap-frozen on dry ice and kept at –80C until used. If the tissue was collected for immunohistochemistry the perfusion of the mice with PBS was followed by perfusion with 4% paraformaldehyde in PBS. Organs were fixed overnight in 4% paraformaldehyde and paraffin embedded. Lewis lung carcinomas were obtained according to previously described methods (Skriver et al. 1984). Skin wounds were made as previously decribed (Rømer et al. 1991). Animal care at the University of Copenhagen and Rigshospitalet was in accordance with national and institutional guidelines.
Antibodies
The following antibodies were purchased from DAKO (Glostrup, Denmark): rabbit anti-von Willebrand Factor (A82), rabbit anti-keratin (Z622), biotinylated swine anti-rabbit (E431), peroxidase-conjugated swine anti-rabbit (P217), alkaline phosphatase-conjugated swine anti-rabbit (D306), and alkaline phosphatase-conjugated biotin–avidin complexes (AP-ABC, K376). Rat monoclonal antibody to mouse macrophages, clone BM-8, was from BMA Biomedicals (Augst, Switzerland). Rat anti-mouse CD34 (clone MEC 14.7) was obtained from Hycult Biotechnology (Uden, Netherlands). The tyramide signal amplification (TSA) kit was purchased from NEN (Boston, MA).
Tissue Extracts
Tissue and cell extracts were prepared as described (Solberg et al. 1992). Briefly, frozen mouse organs were pulverized with a precooled powder pistol followed by acid treatment to remove receptor-bound uPA. Cells and tissue powder were lysed by addition of 1 ml lysis buffer [0.01 M sodium phosphate buffer, pH 7.4, 1% (v/v) Triton X-114, 10 mM ethylenediaminetetraacetic acid (EDTA), 10 μg/ml aprotinin, 1 mM phenylmethylsulfonyl flouride (PMSF)] to 5 × 107 cells or 100 mg tissue powder. The lysates were subjected to temperature-induced phase separation as described (Behrendt et al. 1990).
Chemical Crosslinking Assay and Western Blotting Analysis
Twenty μl detergent-phase extract or cell culture media from transfectants was incubated with either 100 nM mouse ATF (mouse ATF was obtained as described by Solberg et al. 1994) or PBS for 1 hr at 4C. Then 2.5 nM [125I]-human ATF was added to all samples and incubation continued for 1 hr at 4C. Crosslinking was obtained by incubating samples with 10 mM 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) (Pierce; Rockford, IL), and 10 mM N-hydroxysulfosuccinimide (sulfo-NHS) (Pierce) for 20 min at room temperature. The reaction was stopped by adding 10 mM ammonium acetate. Western blotting analysis was made as described (Solberg et al. 1992). Briefly, detergentphase extracts were subjected to SDS-PAGE and electroblotted onto polyvinylidenedifluoride (PVDF) membrane. The membrane was blocked with 1% (w/v) skimmed milk powder in Tris-buffered saline (TBS), followed by incubation with 10 μg/ml preimmune rabbit IgG or 10 μg/ml polyclonal rabbit anti-mouse uPAR IgG. Alkaline phosphatase-conjugated swine anti-rabbit Ig was used for detection by nitroblue tetrazolium/5-bromo-4-chloro-3-indoyl phosphate.
Cloning and Transfection
A fragment of the receptor for mouse urokinase plasminogen activator (smuPAR) cDNA sequence was prepared by standard PCR technique using muPAR cDNA as template (Kristensen et al. 1991a,b). In brief, using Pfu DNA polymerase, a Nhe I site was introduced in front of the noncoding 5′-end of uPAR, and a stop-codon followed by an EcoRI site was introduced at nucleotide position 905 in the 3′-end. The fragment corresponds to amino acids 1–275 of muPAR and gives rise to a soluble form of the receptor by omitting the C-terminal sequence that codes for the GPI anchoring of the protein. The PCR fragment was cut with the appropriate restriction enzymes, and cloned into the high-expression vector PCI (Promega; Madison, WI) under the control of the cytomegalovirus (CMV) immediate-early enhancer/promoter, giving the plasmid PCI–smuPAR. CHO cells were co-transfected with the PCI–smuPAR vector and the PCK–neo vector, and stable transfectants were selected by culturing the cells in medium containing 800 μg/ml G418 (Roche; Basel, Switzerland). When confluent, the clones were kept in serum-free medium for 2 days. The conditioned medium was collected and checked for smuPAR by chemical crosslinking analysis (see above). Clones expressing smuPAR were frozen and used for production of smuPAR.
Purification of smuPAR
Conditioned media from CHO–smuPAR cells grown in 10% fetal calf serum in α-Minimum Essential Medium (MEM) (Gibco; Life Technologies, Roskilde, Denmark) was collected and 0.1 M Tris, pH 8.1, EDTA, and 1 mM PMSF was added before filtration through a 0.8-μm filter. smuPAR was purified by immunoaffinity chromatography using a monoclonal antibody raised against human uPAR in uPAR-deficient mice (KOR1). This antibody recognizes an epitope that is conserved between mouse and human uPAR (unpublished results). The immunoaffinity purification was followed by further purification by reverse-phase HPLC. The purified smuPAR was analysed by SDS-PAGE and the purity was checked by silver staining of the gel.
Specific Polyclonal Rabbit Anti-mouse uPAR Antibodies
Preimmune serum was collected from four individual rabbits. Rabbits were injected SC with 20 μg smuPAR in Freund's complete adjuvant in PBS on Day 0, followed by SC injection of 20 μg smuPAR in Freund's incomplete adjuvant in PBS on Day 18 and every 28 days thereafter. Blood was drawn on Day 28 and every 28 days thereafter. The serum was checked for antibodies specific for mouse uPAR by ELISA. Briefly, ELISA plates were coated overnight with 10 ng/well of purified soluble mouse uPAR. After blocking, the diluted rabbit serum was added to the plates. Bound IgG was detected using peroxidase-coupled swine anti-rabbit antibodies. Color was developed using O-phenylenediamine (OPD) and the optical density measured at 490 nm. The rabbit anti smuPAR IgG was purified using protein G–Sepharose and the concentration of purified IgG was determined using a protein assay from BioRad (Copenhagen, Denmark).
Immunohistochemistry
Paraffin sections were deparaffinized in xylene, digested with 0.03% trypsin for 10 min at 37C, blocked in 5% swine serum in TBS with 0.25% bovine serum albumin (BSA) (TBS–BSA), and incubated with 10 μg/ml polyclonal rabbit IgG in TBS-BSA raised against soluble mouse uPAR. Then biotinylated swine anti-rabbit was applied, followed by AP–ABC according to the manufacturer's instructions. Slides were developed for 10–30 min. with Fast Red and counter-stained with Mayer's hematoxylin. For signal amplification with biotinyl tyramide (Bobrow et al. 1989), detection with biotinylated detecting antibodies was followed by horseradish peroxidase-coupled streptavidin, amplification with biotinyl tyramide for 10 minutes, and again horseradish peroxidase-coupled streptavidin. Finally, sections were developed for 10 min with NovaRed substrate (Vector Laboratories; Burlingname, CA) according to the manufacturer's instructions.
As negative controls, we used preimmune rabbit IgG, rabbit anti-mouse uPAR IgG preincubated with 10-fold molar excess of smuPAR, and tissues from uPAR-deficient mice.
Results
Organ distribution of mouse uPAR
To determine the level of uPAR expression in mice under normal physiological conditions, extracts were prepared from various tissues of BALB/C mice. Detergent-phase extracts were analyzed by a chemical crosslinking method based on the ability of EDC/NHS to covalently crosslink 125I-labeled human ATF to mouse uPAR (Solberg et al. 1992). The specificity of this detection method was verified by the use of unlabeled mouse ATF as a competitive inhibitor of the crosslinking reaction. No crosslinked product was obtained after preincubation with 100 nM mouse ATF. The highest yield of crosslinked product was found in lysates from spleen, lung, and kidney (Figure 1, Lanes 1–6) and in lysates from primary tumors of the highly metastatic Lewis lung carcinoma that expressed high levels of uPAR (Figure 1, Lanes 7 and 8), whereas no uPAR was detected in lysates of brain, heart and liver tissues (Figure 1, Lanes 9–14).
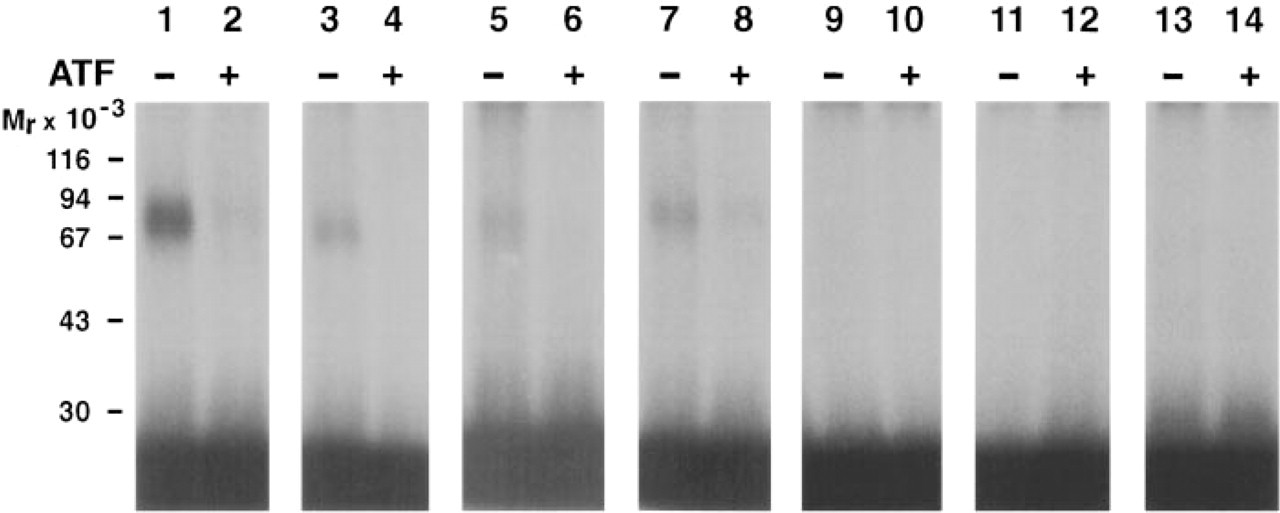
Detection of mouse uPAR in tissues by chemical crosslinking. Tissue extracts prepared either from spleen (Lanes 1,2), lung (Lanes 3,4), kidney (Lanes 5,6), Lewis lung carcinoma Lanes 7,8), heart (Lanes 9,10), liver (Lanes 11,12), or brain (Lanes 13,14) were incubated with 2.5 nM 125I-labeled human ATF and crosslinked using EDC-NHS. Samples marked with + were preincubated with 100 nM mouse ATF. All samples were subjected to SDS-PAGE in 10% polyacrylamide gels, followed by autoradiography.
Generation of Polyclonal Rabbit Antibodies to Mouse uPAR
To study the histological localization of uPAR in the mouse, polyclonal antibodies against mouse uPAR were generated by immunization of rabbits using recombinant mouse uPAR as antigen. Soluble mouse uPAR was produced by transfection of CHO cells with a construct encoding a soluble form of mouse uPAR. Purification of smuPAR from conditioned media was done by immunoaffinity chromatography, using a monoclonal antibody (KOR 1) that binds to an epitope conserved between mouse and human uPAR. The immunoaffinity purification was followed by a further purification by reverse-phase HPLC. The purity and electrophoretic mobility of the mouse uPAR antigen were monitored by SDS-PAGE and silver staining (Figure 2, Lane 1). To test whether the recombinant smuPAR was folded correctly, its ligand-binding properties were examined by surface plasmon resonance using an optical biosensor (Biacore 2000), as described previously for the human protein (Gårdsvoll et al. 1999). Real-time binding kinetics using purified smuPAR coupled to the sensor chip revealed a high-affinity interaction between immobilized smuPAR and mouse uPA, whereas human uPA exhibited only weak affinity (data not shown). This apparent species specificity of the interaction between purified immobilized and mouse uPA vs human uPA is in complete agreement with data obtained by others (Estreicher et al. 1989; Tressler et al. 1999).
The purified smuPAR preparation was then used as antigen for immunization of four rabbits and serum was tested for specific IgG by ELISA. Specificity of the purified IgG from these rabbits was subsequently examined by Western blotting analysis of detergent phase extracts of Lewis lung carcinoma (Figure 2, Lane 2), spleen (Figure 2, Lanes 3 and 4), and lung (Figure 2, Lanes 5 and 6) from wild-type (Figure 2, Lanes 3 and 5) or uPAR-deficient (Figure 2, Lanes 4 and 6) mice. A single band corresponding to the migration of purified smuPAR was seen in extacts from Lewis lung carcinoma and wild-type spleen and lung, whereas no bands were observed in extracts prepared from uPAR-deficient mice. No bands were obtained when preimmune IgG from the same rabbit was used (data not shown).
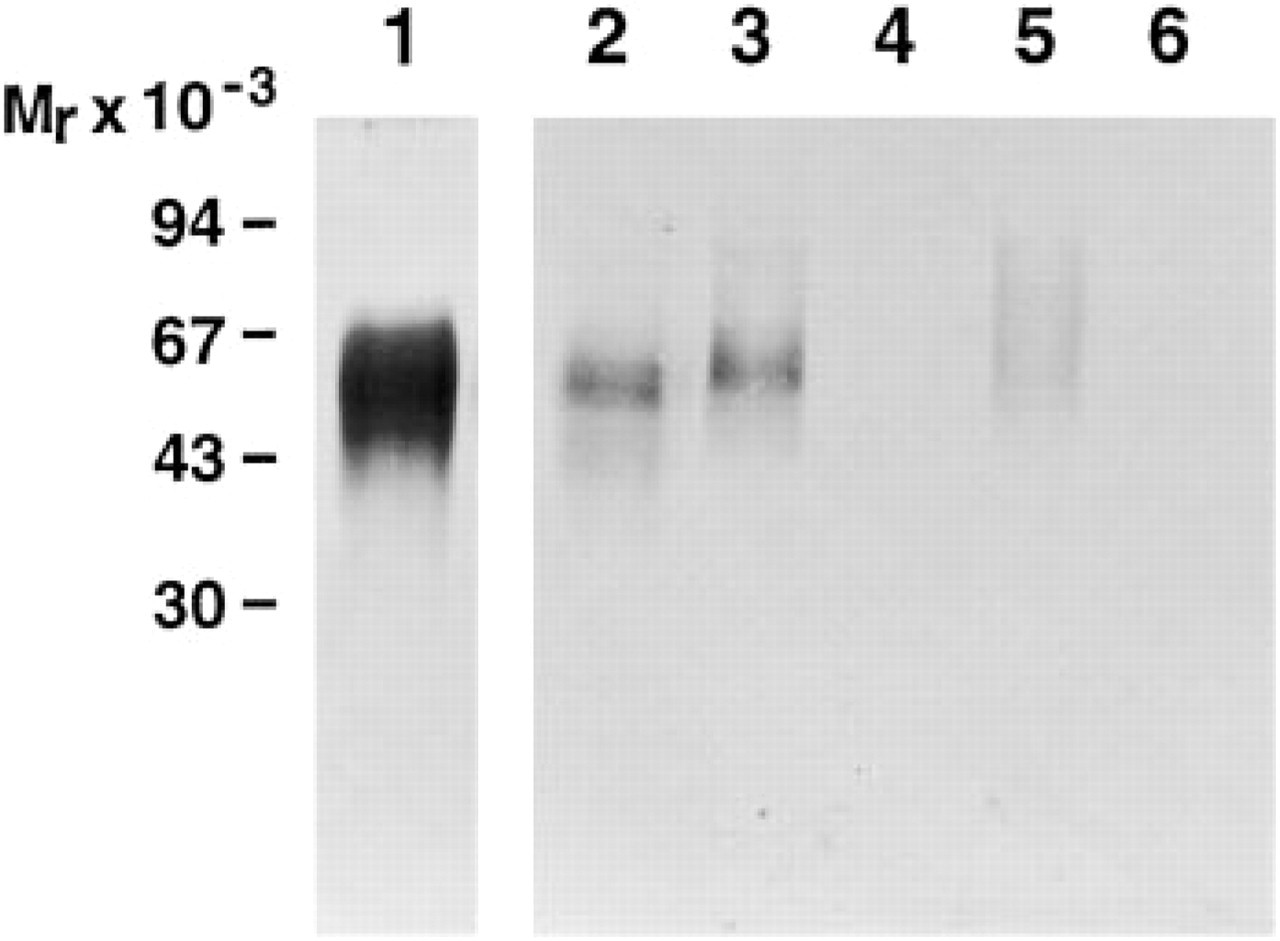
Detection of mouse uPAR by immunoblotting. Silver staining of purified soluble mouse uPAR (2 μg) after SDS-PAGE in a 10% polyacrylamide gel (Lane 1). Western blotting analysis of uPAR in extracts of Lewis lung carcinoma (Lane 2), spleen (Lanes 3,4), and lung tissues (Lanes 5,6) from uPAR wild-type (Lanes 3,5) and uPAR-deficient mice (Lanes 4,6), using 10 μg/ml rabbit anti-muPAR as the primary antibody.
Histological Localization of uPAR in Organs From Healthy Mice
Immunohistochemistry was used to map the histological localization of uPAR protein under normal and pathological conditions in the mouse. For all organs examined, tissues from at least two animals were investigated, giving similar results. A tissue section from each organ was stained with polyclonal rabbit antibodies against cytokeratin to ensure that proteins were intact after fixation and embedding of the tissue, as judged by positive staining of keratin in epithelial cells. Equivalent tissue from mice rendered uPAR-deficient by homologous recombination (Bugge et al. 1995) served as a unique control for the staining of uPAR obtained in normal mouse tissue with the polyclonal rabbit anti-mouse uPAR IgG. In addition, more conventional negative controls using preimmune IgG also revealed no staining when applied at the same concentration on tissue from normal mice.
Immunohistochemistry of lung tissue showed uPAR staining in the alveoli and pulmonary vessels, whereas the bronchial epithelium was negative (Figure 3A). No staining was seen with either preimmune rabbit IgG on wild-type lung tissue (Figure 3B) or the polyclonal immune IgG on equivalent lung tissue obtained from uPAR-deficient mice (Figure 3C). By evaluation of adjacent sections of lung tissue stained with antibodies against von Willebrand factor, cytokeratin, and macrophage clone BM-8, it was evident that endothelial cells of the alveoli express uPAR immunoreactivity, whereas we were unable to clearly identify immunostaining of pneumocytes and alveolar macrophages (data not shown).
In the kidney, the highest intensity of uPAR immunoreactivity was observed in glomeruli and in vessels in the medulla. Vessels in the cortex showed weaker uPAR staining (Figure 3D). Immunohistochemical staining with an antibody against CD34, an endothelial cell marker, showed a staining pattern indistinguishable from that obtained with uPAR-specific antibodies, suggesting that all endothelial cells of the kidney express uPAR. No uPAR immunoreactivity was seen in kidneys from uPAR-deficient mice (Figure 3E).
In the spleen, uPAR immunoreactivity was seen in the perifollicular zone, an area that contains a high number of leukocytes with a cell type composition similar to that found in peripheral blood (Figure 3F). In the liver, uPAR immunoreactivity was detected only in the arteries and arterioles. No other cells were positive, including veins, sinosoids, and Kuppfer cells (data not shown). In the heart, uPAR was detected in the endothelial cells of arteries, whereas capillaries showed little or no immunoreactivity. CD34 immunostaining demonstrated the presence of multiple capillaries that were generally devoid of uPAR immunoreactivity (data not shown).
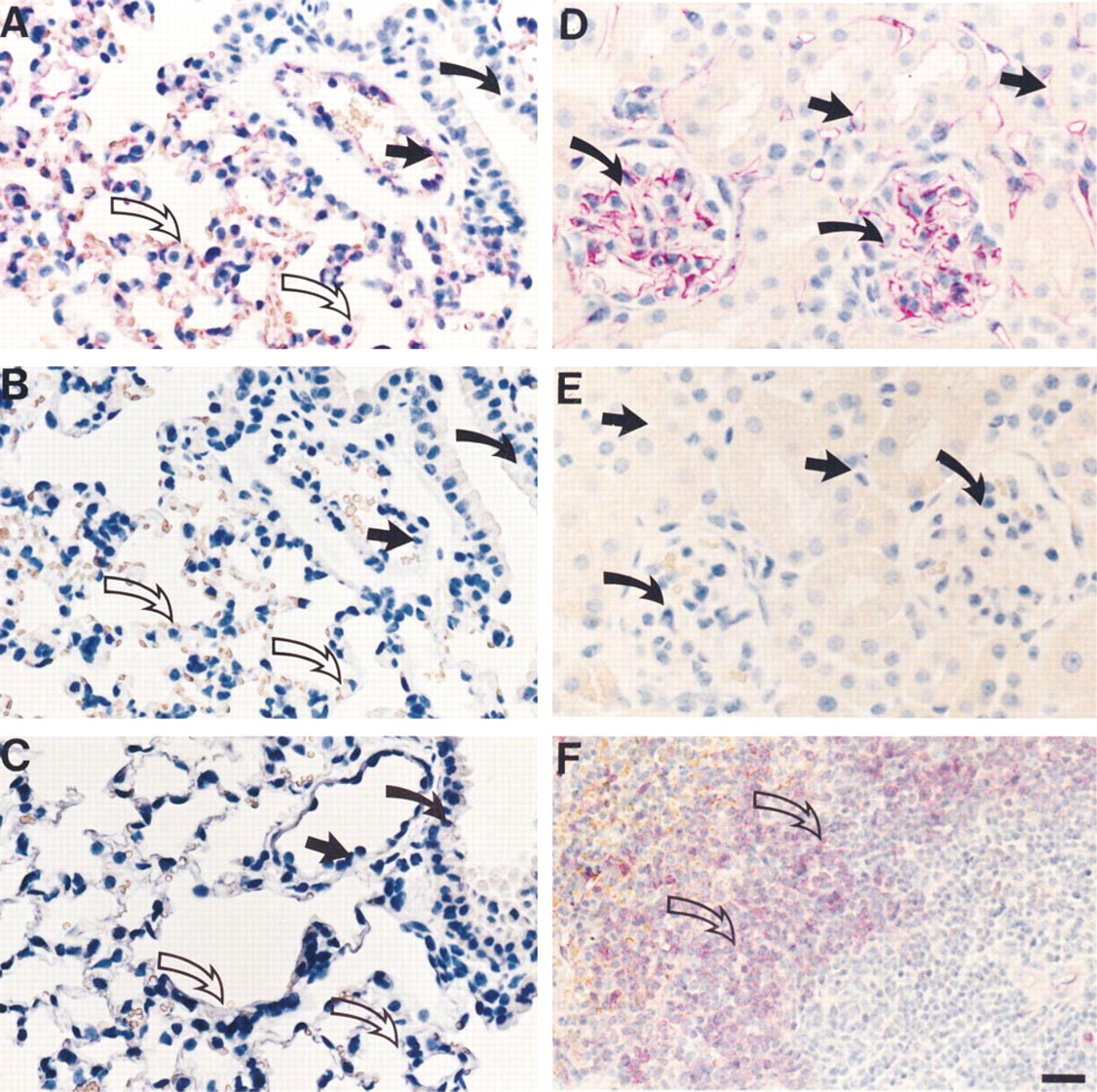
Immunohistochemical localization of uPAR in normal lung, kidney, and spleen tissue. (
As a common denominator, pronounced uPAR immunoreactivity was consistently observed in normal mouse organs undergoing a defined tissue remodeling process. First, sections of 16-hr-old mouse skin wounds showed strong uPAR reactivity in the moving keratinocytes at the edge of the wound, whereas the non-migrating keratinocytes were negative. uPAR immunoreactivity was also seen in granulocytes infiltrating the area just below the wound crush. Endothelial cells in the wound area and the nearby normal skin stained positive for uPAR protein (Figure 4A). Second, during late pregnancy, spongiotrophoblast cells in the placenta showed distinct uPAR immunoreactivity localized to the plasma membrane. Endothelial cells in the placenta stained positive for uPAR (Figure 4B), as did the decidual cells bordering the uterine epithelium (data not shown). Third, in post-lactational involuting mammary glands, weak to moderate staining could be detected in the regressing glandular tissue (Figure 4C). Strong uPAR immunoreactivity was also seen in granulocytes localized in the lymph nodes of the mammary gland and in cells, probably macrophages, present in the subcapsular sinuses of the lymph nodes (data not shown). For all three tissues undergoing tissue remodeling, no uPAR immunoreactivity was seen when equivalent tissues obtained from uPAR-deficient mice were stained with the same rabbit anti-mouse uPAR IgG (Figures 4D–4D).
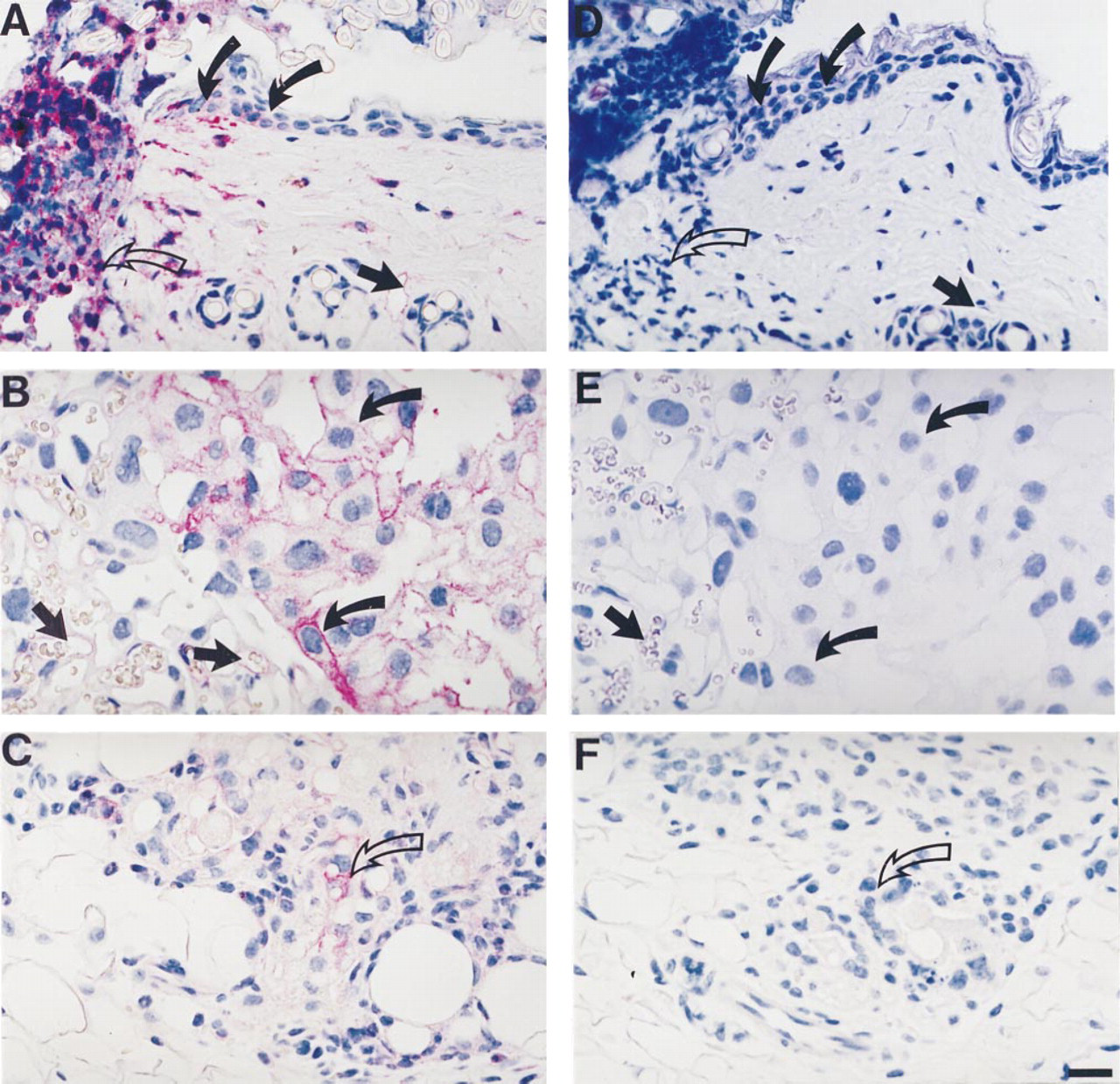
Localization of uPAR immunoreactivity in skin wounds, placenta, and mammary gland involution. (
Lewis Lung Carcinoma
Primary tumors of Lewis lung carcinoma showed clear surface-localized uPAR immunoreactivity (Figure 5A). There was pronounced heterogeneity in the staining of parts of individual tumors: some parts stained strongly, whereas other parts were completely devoid of uPAR immunoreactivity. In addition, vessels and neutrophils infiltrating the necrotic foci of the tumor showed uPAR immunoreactivity (Figure 5A). Metastases in the lung also showed uPAR immunoreactivity (data not shown). As control for the staining specificity, rabbit anti-mouse uPAR IgG was incubated with excess antigen before immunostaining. After antigen absorption of the antibody (Figure 5B) and with preimmune IgG (data not shown), no staining of the tumor was seen.
Discussion
uPAR has been implicated in ECM degradation under normal physiological processes that involve tissue remodeling and during cancer cell invasion and metastasis. Mice rendered uPAR-deficient by gene targeting (Bugge et al. 1995) may provide a useful tool to establish the function of uPAR in vivo. To reveal abnormalities in uPAR-deficient mice, a precise knowledge of uPAR expression in vivo in normal mice is required. The majority of the current knowledge regarding uPAR expression and function is based on studies using cultured cells in vitro, which suffer from obvious limitations compared to in vivo expression. Tissue and cell distribution of defined proteins in vivo can be determined by immunohistochemistry, provided that antibodies with high affinity and specificity are available. However, unambiguous interpretations of such immunohistochemical stainings are often hampered by the lack of appropriate controls for specificity of the staining obtained. In the present study, tissues from mice genetically deficient in uPAR served as crucial controls for the specificity of the uPAR immunoreactivity. Consequently, the data presented here on uPAR protein expression in normal mouse organs, as well as in mouse tissues undergoing extensive tissue remodeling, can be interpreted with great certainty, even for tissue sections in which only a moderate level of uPAR staining is detected.
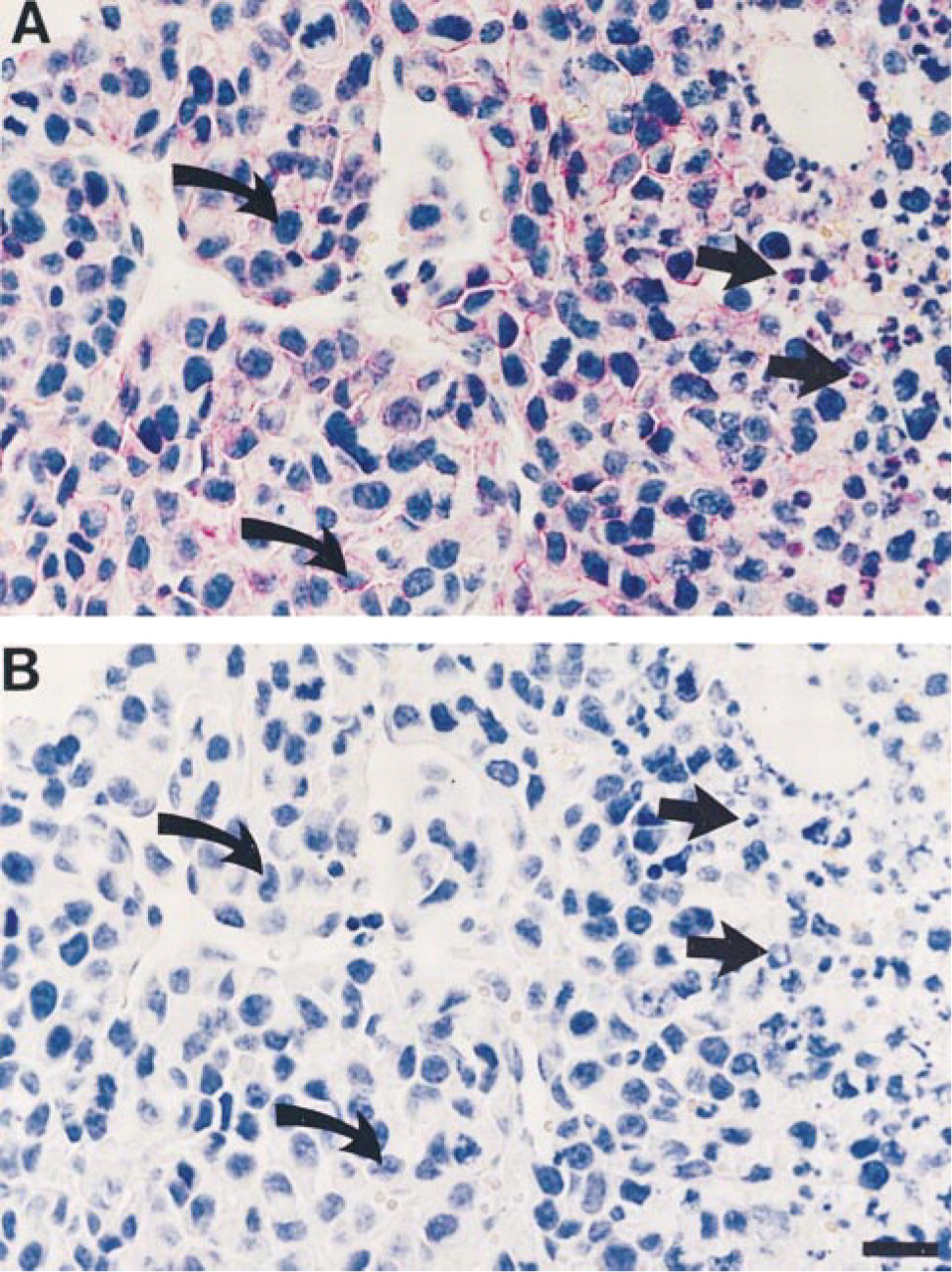
Localization of uPAR immunoreactity in Lewis lung carcinoma. (
Immunoreactivity for uPAR was detected throughout the alveoli of the lungs. This finding is consistent with the relatively high level of uPAR protein detected in extracts of lung tissue by chemical crosslinking. Because of the rich capillary network, it was difficult to rigorously conclude that pneumocytes and macrophages are actually responsible for some of the uPAR staining. Immunohistochemical double labeling is required to clarify a possible contribution from such cells. In situ hybridizations on lung tissue have thus far detected uPAR mRNA only in a few scattered cells bordering the alveolar space, possibly representing alveolar macrophages (Almus–Jacobs et al. 1995). The apparent lack of mRNA for uPAR in the endothelial cells of the lung compared to the positive immunohistological staining might be due to a low level of uPAR mRNA (below the detection limit for in situ hybridization) combined with a long half-life of the uPAR protein on the membrane of these cells.
Crosslinking analysis revealed appreciable amounts of functional uPAR protein in the kidneys, an organ that has previously been shown to contain high amounts of uPA (Larsson et al. 1984; Kristensen et al. 1991a,b). Intriguingly, uPA was primarily located in the proximal and distal tubules of the outer part of the medulla, whereas uPAR immunoreactivity was confined to glomeruli and vessels of the medulla and the cortex. No co-localization of uPA and its receptor was therefore seen in the kidney, which raises the intriguing possibility that uPAR serves other functions in the kidney than merely confining uPA to the cell surface.
Peripheral blood leukocytes express uPAR, and it has previously been reported that the cell surface expression of uPAR rapidly increases due to a translocation of a certain subset of granules after leukocyte activation (Nykjár et al. 1990, 1992; Ploug et al. 1992; Plesner et al. 1994b). This finding is consistent with the present detection of a relatively high level of uPAR protein in extracts of spleen by chemical cross-linking. Furthermore, immunohistochemistry demonstrated uPAR expression in the perifollicular zone of the spleen, an area that contains considerable numbers of leukocytes with a cell type composition similar to that found in peripheral blood. These findings are supported by data obtained by others using Northern blotting analysis and in situ hybridization (Almus-Jacobs et al. 1995). uPAR has been found in close association with β2-integrins on leukocytes (Xue et al. 1994; Bohuslav et al. 1995), and recruitment of leukocytes into the peritoneum by thioglycollate induction has been reported to be significantly reduced in uPAR-deficient mice compared to wild-type animals (May et al. 1998).
The confinement of uPAR immunoreactivity to the migrating keratinocytes of incisional skin wounds is in complete accordance with in situ hybridization for uPAR mRNA (Rømer et al. 1991). Pronounced uPAR immunoreactivity was also observed for granulocytes infiltrating the wound area; in contrast, these cells did not contain detectable levels of uPAR mRNA as probed by in situ hybridization (Rømer et al. 1991). The apparent discrepancy between these data can be resolved by the fact that uPAR protein is stored in cytoplasmic granules of resting neutrophils and that these granules can be released and uPAR translocated to the cell surface after mild stimulation (Plesner et al. 1994a). Therefore, no de novo protein synthesis is required for activated neutrophils in the wound crush to express uPAR, which may account for the lack of detectable mRNA. A similar phenomenon has been reported for neutrophils in human colon cancer (Pyke et al. 1994).
Involuting mammary glands reveal uPAR immunoreactivity in the regressing glandular tissue, which also expresses uPA (Larsson et al. 1984). This may be related to the recent finding that mammary gland involution is delayed in plasminogen-deficient mice (Lund et al. 2000).
During late pregnancy, uPAR immunoreactivity was localized to spongiotrophoblast cells of the placenta, as well as in the decidual cells bordering the uterine epithelium. The decidual cells have previously been shown also to possess uPA reactivity (Larsson et al. 1984). In contrast, no detectable amounts of uPA or uPAR mRNA was found in the placenta at late pregnancy by in situ hybridization. In early pregnancy (11.5 days p.c.), however, uPAR mRNA was detected in both spongiotrophoblast cells and mesometrial decidua (Teesalu et al. 1998).
uPAR and uPA have previously been detected only in activated endothelial cells in vivo during neovascularization or inflammation (Bacharach et al. 1992; Pepper et al. 1993). However, in the present study we have consistently detected uPAR immunoreactivity in quiescent endothelial cells in many organs of the normal mouse, e.g., lung, kidney, placenta, normal skin, liver, heart, uterus, testis, urinary bladder, and thymus. This finding is also contradictory to data obtained by in situ hybridization in which endothelial cells were found positive for uPAR mRNA only after LPS treatment (Almus–Jacobs et al. 1995). As mentioned above, one likely explanation for this discrepancy is that there is a low level of uPAR mRNA (below the detection limit for in situ hybridization), combined with a long half-life of uPAR protein, on the membrane of endothelial cells. Furthermore, the use of uPAR-deficient tissues as negative controls allows fairly weak staining signals to be considered uPAR-positive. The finding of uPAR in the endothelium is not simply a reflection of the developmental stage of the mice, because the uPAR immunostaining pattern of lungs from mice 8–12 weeks of age was indistinguishable from the staining obtained in lungs from mice 10 months of age. No uPA protein nor mRNA has been detected in quiescent endothelial cells in the normal mouse (Larsson et al. 1984; Kristensen et al. 1991b).
The highly metastatic Lewis lung carcinoma showed distinct uPAR staining localized to the surface of the tumor cells in both primary tumor and metastases of the lung. However, the staining in the primary tumor was very heterogeneous, showing some areas with pronounced uPAR immunoreactivity whereas others were totally devoid of staining. Although all Lewis lung cells in culture probably express uPAR, the lack of expression in some of the tumor cells in vivo is probably not the result of downregulation. Rather, the expression of uPAR at the invasive front is likely the result of upregulation of uPAR induced by potential signal(s) from the stroma. By analogy, only cancer cells located at the invasive front in human colon cancer express uPAR, whereas many colon cancer cell lines uniformly express uPAR in vitro (Pyke et al. 1994). A similar localization of uPA immunoreactivity in Lewis lung carcinoma has been reported previously (Skriver et al. 1984), which was mainly confined to areas with local invasion. Many cancer cells have previously been shown to express uPAR in vitro (Nielsen et al. 1988) and in vivo (Pyke et al. 1991, Rømer et al. 1994). It has been proposed that uPAR plays a role during invasion and metastasis of cancer cells (Danø et al. 1994), and several studies using in vivo model systems combined with antagonists of the uPA–uPAR interaction substantiate this hypothesis (Crowley et al. 1993; Kook et al. 1994; Min et al. 1996; Kim et al. 1998). Measurements of uPAR levels in extracts of tumor tissues have furthermore demonstrated a correlation between high levels of uPAR protein in tumor tissue and poor patient survival (Ganesh et al. 1994; Pedersen et al. 1994; Grøndahl–Hansen et al. 1995).
The generation of uPAR-deficient mice (Bugge et al. 1995), in combination with the immunohistochemical data of the distribution of uPAR protein in the mouse, will help to elucidate the functional role of uPAR in vivo under both normal and pathological conditions.
Footnotes
Acknowledgments
Supported by the Danish Cancer Society and the Danish Research Council.
We thank Dr Thomas Bugge for the uPAR-deficient mice. We are grateful to Anette Bartels, Pia Gottrup Knudsen, and Helle Malmstedt for excellent technical assistance.