
Abstract
The newly developed Animal Research Kit (ARK) offers a simple and economic way of biotinylating mouse primary antibodies for background-free immunostaining of mouse and rat tissue specimens. Biotinylation involves the use of a biotinylated goat anti-mouse immunoglobulin Fab fragment mixed with a mouse primary antibody and subsequent blocking with normal mouse immunoglobulin. Because a reliable immunoenzyme double staining procedure on human tissue specimens with two unlabeled mouse primary antibodies of identical subclass is almost impossible, we have tested the performance of ARK biotinylation of one primary antibody in a multistep indirect/direct staining protocol. The multistep double staining procedure involved the subsequent application of an unlabeled mouse monoclonal antibody (MAb) 1 detected with an enzyme-labeled EnVision reagent, normal mouse serum for blocking, followed by a biotinylated mouse MAb 2 and enzyme-labeled streptavidin. Alkaline phosphatase and peroxidase enzymatic activities were developed last. Double staining results obtained with an ARK biotinylated reagent were compared with a truly biotinylated reagent using N-hydroxy succinimide–biotin for conjugation. It appeared that both biotinylation procedures revealed identical double staining results. Although a limited number of antibody combinations have been tested, it is clear that this double staining procedure will be successful for many antibody pairs.
Keywords
I
The Animal Research Kit (ARK) was originally designed for background-free immunohistochemical staining with mouse primary antibodies on tissue specimens from any species, including mouse and rat. The procedure is based on in vitro labeling of a mouse primary antibody with a biotinylated anti-mouse Fab fragment, resulting in biotinylation of the primary antibody. Remaining biotinylated anti-mouse Fab fragments are subsequently blocked with normal mouse immunoglobulins (Tuson et al. 1990). After the successful application of ARK biotinylated antibodies for human tissue specimens was feasible (H Göbel, personal observation), the idea arose to employ this way of biotinylation for an indirect-direct multistep double staining procedure (Van der Loos et al. 1987, 1989) according to the scheme in Figure 1. For detection of the first unlabeled primary antibody, a non-streptavidinbiotin-based two-step EnVision/goat anti-mouse, horseradish peroxidase (HRP), or EnVision/goat anti-rabbit + mouse, alkaline phosphatase (AP) (Bisgaard and Pluzek 1996) technique was used. The EnVision polymer technique yields a staining efficiency comparable to that of a three-step streptavidin-biotin method (Sabattini et al. 1998; Vyberg and Nielsen 1998). After blocking of all free anti-mouse binding sites of the EnVision reagent with normal mouse serum (Van der Loos et al. 1987), a second primary antibody was applied in a biotinylated format. Depending on the choice for the enzymatic label for the EnVision reagent, the double staining procedure was finished either with the application of streptavidin/AP or streptavidin/HRP. The enzymatic activities of AP and HRP were developed in this order in blue and red respectively.
In this study we have compared on serial sections the use of an ARK-biotinylated reagent with a “true” N-hydroxy succinimide-biotin conjugate. After observing no difference between the ARK biotin reagent and “true” biotin conjugate, six straightforward mouse primary antibody combinations with identical IgG subclass were tested on cryostat and paraffin-embedded tissue specimens, including the application of heat-induced antigen retrieval (Shi et al. 1997).
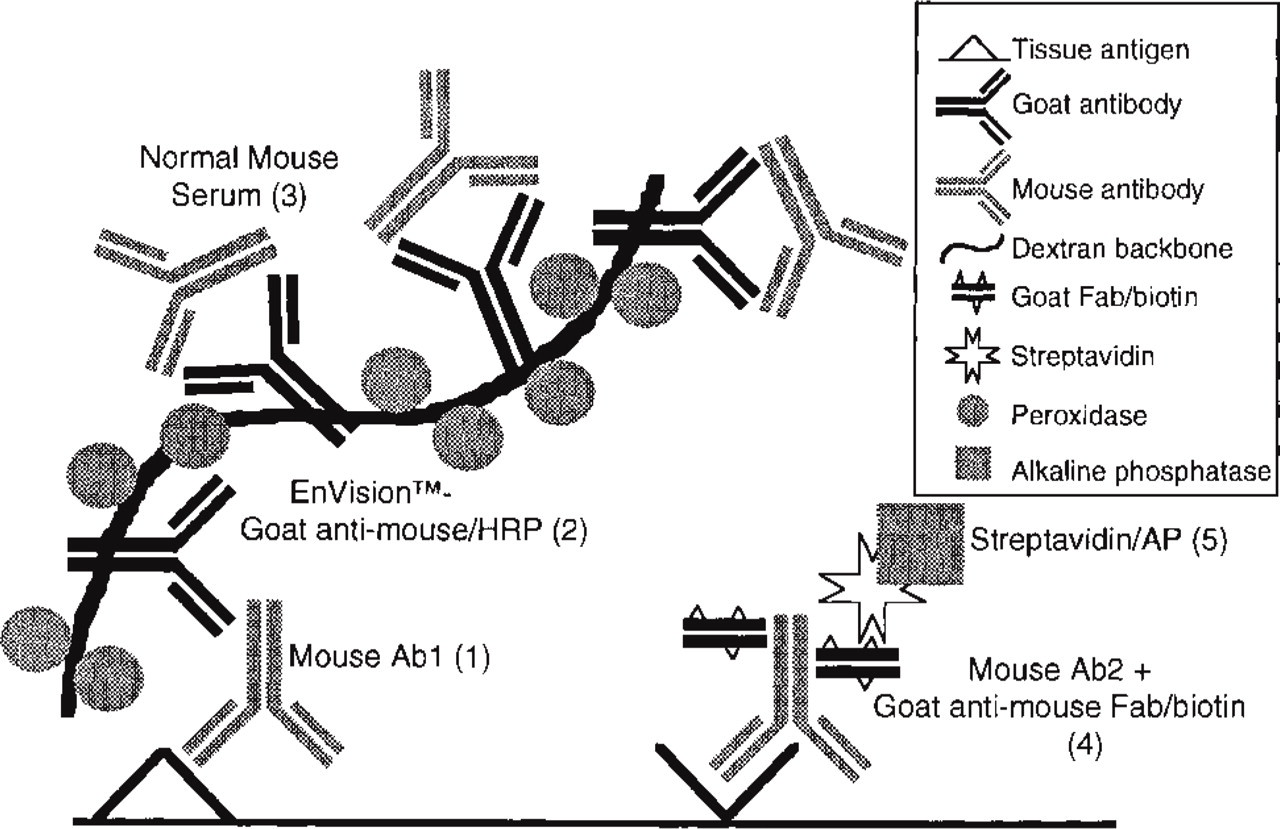
Schematic representation of the multistep double staining procedure using an unlabeled primary antibody and an ARK–biotinylated reagent. Figures in parentheses refer to step numbers in Table 2.
Materials and Methods
Tissue Preparation
The following human tissue specimens were used for cryostat sections: two tonsils with nonspecific changes obtained at surgery from young children and two aortic segments with atherosclerotic lesions obtained at autopsy. Tissue samples were snap-frozen in liquid nitrogen and stored at −80C. Six-μm cryostat serial sections were cut and dried overnight under a fan. Tissue sections were numbered and stored dry at −80C. On use, the sections were fixed in pure acetone (10 min, 4C) and briefly air-dried. Endogenous peroxidase activity was blocked with 0.1% sodium azide + 0.3% peroxide in 50 mM Tris-HCl-buffered saline, pH 7.8 (TBS; 20 min, room temperature) (Li et al. 1987), followed by TBS washing. The following human tissue specimens were formalin-fixed (approximately 48 hr) and paraffin-embedded: one skin melanoma, two breast carcinomas obtained at surgery, and two carotid arteries with atherosclerotic lesions obtained at autopsy. Six-μm paraffin sections were cut and rehydrated in xylene and graded alcohols. Endogenous peroxidase activity was blocked with 0.3% peroxide in methanol (20 min, room temperature) and washed three times with distilled water.
Reagents
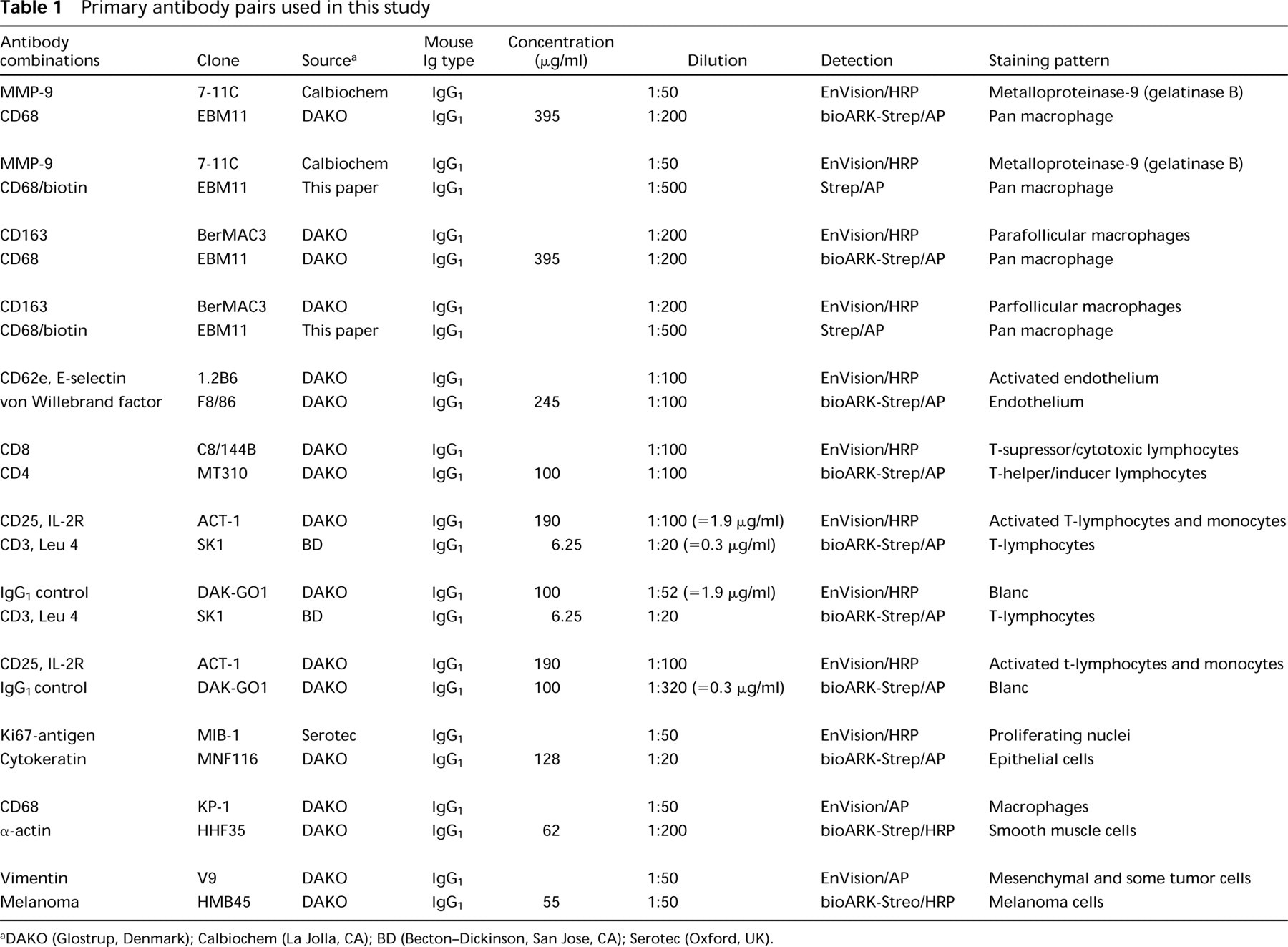
Primary antibody pairs used for double staining are listed in Table 1. EnVision/goat anti-mouse IgG, HRP (EnVision/GAM-HRP), EnVision/goat anti-mouse + rabbit IgG, AP (EnVision/GAM + GAR-AP), normal mouse serum, normal goat serum, AP-conjugated streptavidin, HRP-conjugated streptavidin, and ARK kit (consisting of a biotinylation reagent and a blocking reagent; other kit components were not used in this study) were from DAKO (Glostrup, Denmark).
CD68, EBM11/biotin
The IgG1 fraction from 10 ml anti-CD68, EBM11 was purified using a Agarose-protein A column and buffers from Pierce (Rockford, IL), according to the instructions of the manufacturer. The yield was 1.3 mg of purified anti-CD68, EBM11 in 1.5 ml Tris-HCl (50 mM, pH 7.8) stored at −80C in 500-μl aliquots. One aliquot (= 430 μg) was biotinylated with ANHS-biotin ester according to the instructions from the manufacturer (Roche/Boehringer), using a molar ratio of biotin ester and immunoglobulin 10:1. After biotinylation, the conjugate was tested immunohistochemically by applying a 60-min incubation at room temperature. AP-conjugated streptavidin was used as second step, and Fast Blue BB as chromogen. The optimal dilution of the CD68, EBM11/biotin conjugate was found near 1:200.
Primary antibody pairs used in this study
aDAKO (Glostrup, Denmark); Calbiochem (La Jolla, CA); BD (Becton–Dickinson, San Jose, CA); Serotec (Oxford, UK).
In Vitro Biotinylation with ARK
The DAKO ARKulator (cat. no. S3953), a software application based on Microsoft Excel, was used for calculation of the reagent volumes needed for in vitro biotinylation. The regular working dilution of the antibody (equal to the dilution used for a regular three-step streptavidin detection technique), specific mouse immunoglobulin concentration of the antibody, and desired working volume were input data, and the volumes of primary antibody, buffer, biotinylation reagent and blocking reagent were read-outs. The in vitro biotinylation of the second mouse MAb with biotinylated goat anti-mouse Fab fragments was done according to the instructions with the ARK reagents. For example, for the anti-CD68, EBM11 antibody, 2.5 μl of antibody (395 μg/ml specific IgG) was first diluted in 468 μl TBS, and 10 μl biotinylation reagent (goat anti-mouse Fab/biotin) was added. After incubation for 15 min, 20 μl of blocking reagent (normal mouse immunoglobulin) was added to block the unbound biotinylation reagent (5 min). The reagent was now ready for use. Input data of the other primary antibodies were based on the dilution and IgG concentration as presented in Table 1.
Double Staining Procedure
Antisera and antibody-enzyme conjugates were diluted in TBS + 1% bovine serum albumin (BSA). TBS washings were performed between all steps (three times for 2 min), and all incubations were performed at room temperature unless otherwise stated. Normal goat serum was applied first for blocking nonspecific binding. The multistep double staining technique was performed as previously described (Van der Loos et al. 1993; Van der Loos 1999). The subsequent incubation steps can be found in Table 2. Dilution of the primary antibodies and the applied detection system are shown in Table 1. Figure 1 represents a scheme of the double staining procedure.
Multistep immunoenzyme double staining protocol combining one unlabeled and one ARK-biotinylated primary antibody

aWhen the primary antibody 1 provides strong and intense staining, it is optional to replace Steps 2 and 5 by EnVision/GAM + GAR/AP (undiluted) and streptavidin/HRP (1:400), respectively.
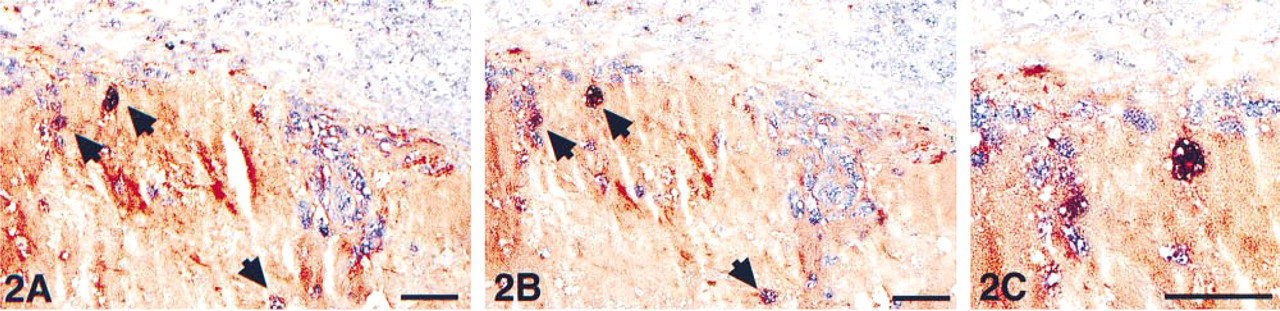
Acetone-fixed cryostat section from an aortic segment, showing a detail of the intima of an atherosclerotic lesion. Immunohis-tochemical double staining was performed with anti-CD68 marking macrophages in blue (AP) and anti-MMP-9 marking the gelatinase A degrading enzyme in red (HRP). Gelatinase A is abundantly present in the extracellular space in this lipid-rich atherosclerotic plaque. A few macrophages appear in a purple mixed color, indicating co-localization (arrowheads). (
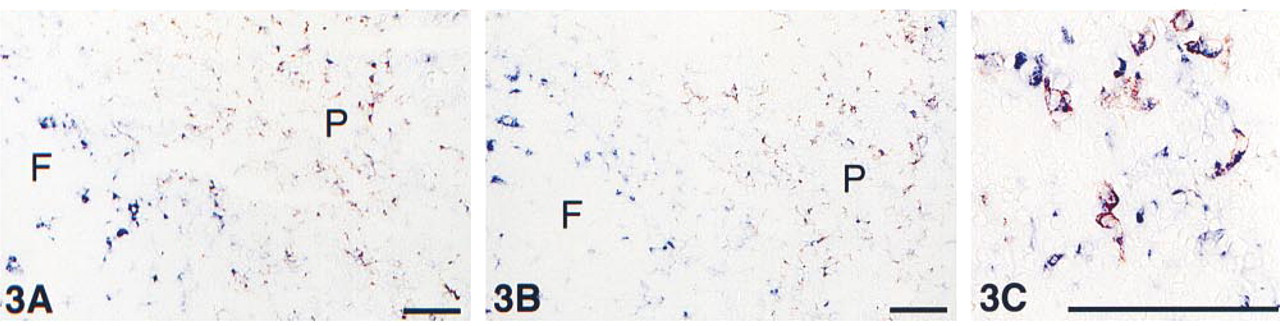
Acetone-fixed cryostat section from tonsil, showing an overview of a follicle center (F) and adjacent parafollicular area (P). Immunohistochemical double staining was performed with anti-CD68 marking macrophages in blue (AP, cytoplasmic staining) and anti-CD163, BerMAC3 marking activated macrophages in red (HRP, membrane staining). The macrophages in the parafollicular area are mostly double stained whereas the “starry sky” macrophages in the follicle center are all single blue. Because of separate cell compartments, a purple mixed color is absent. (
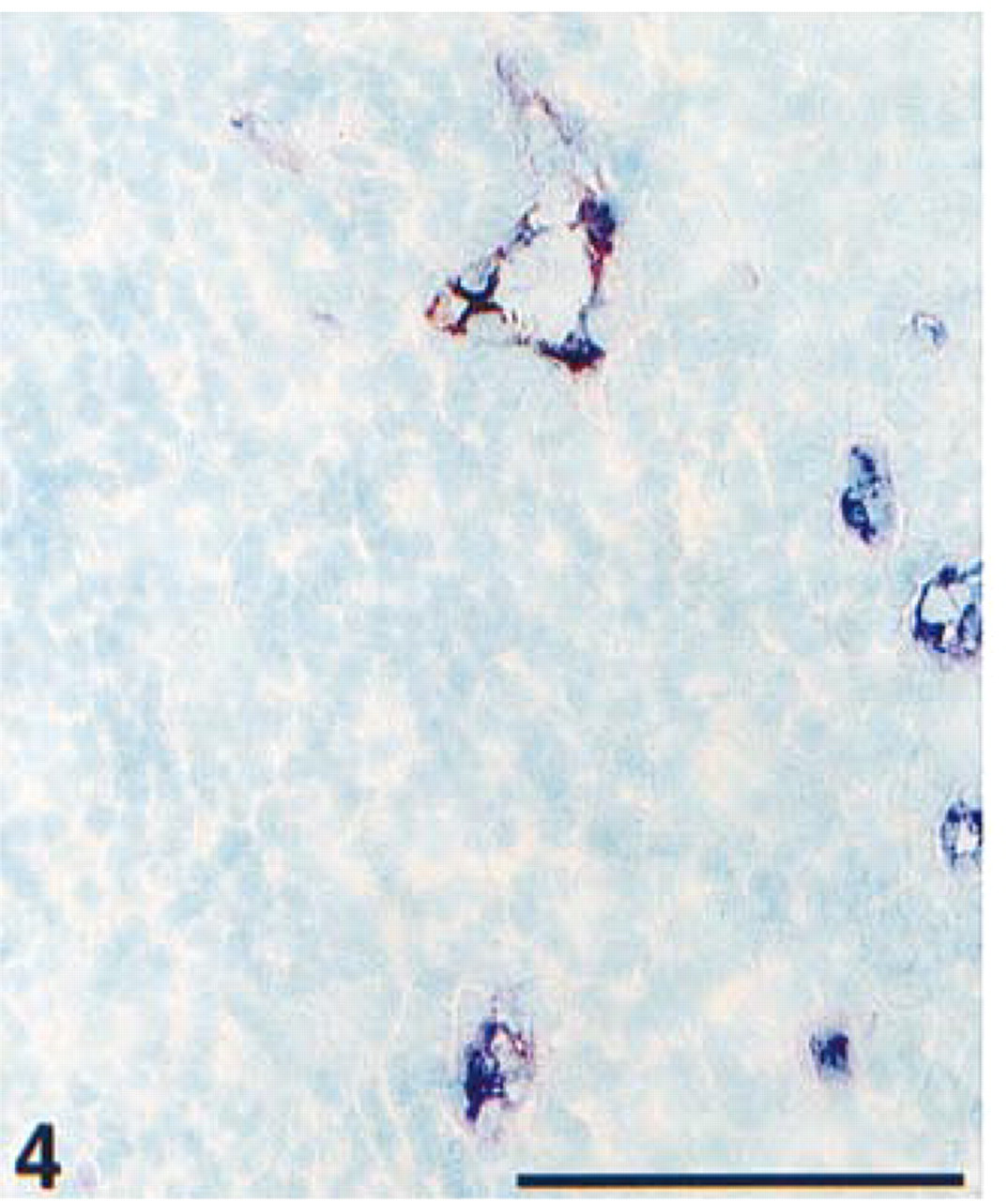
Detail of an acetone-fixed cryostat section from tonsil. Immunohistochemical double staining was performed with ARK-biotinylated anti-von Willebrand factor in blue (AP) and anti-CD62e, E-selectin in red (HRP). Normal endothelial cells stain single blue; activated endothelial cells at sites of inflammation with upregulated CD62e show a purple mixed color. Nuclear counterstain with methyl green. Bar = 100 μm.
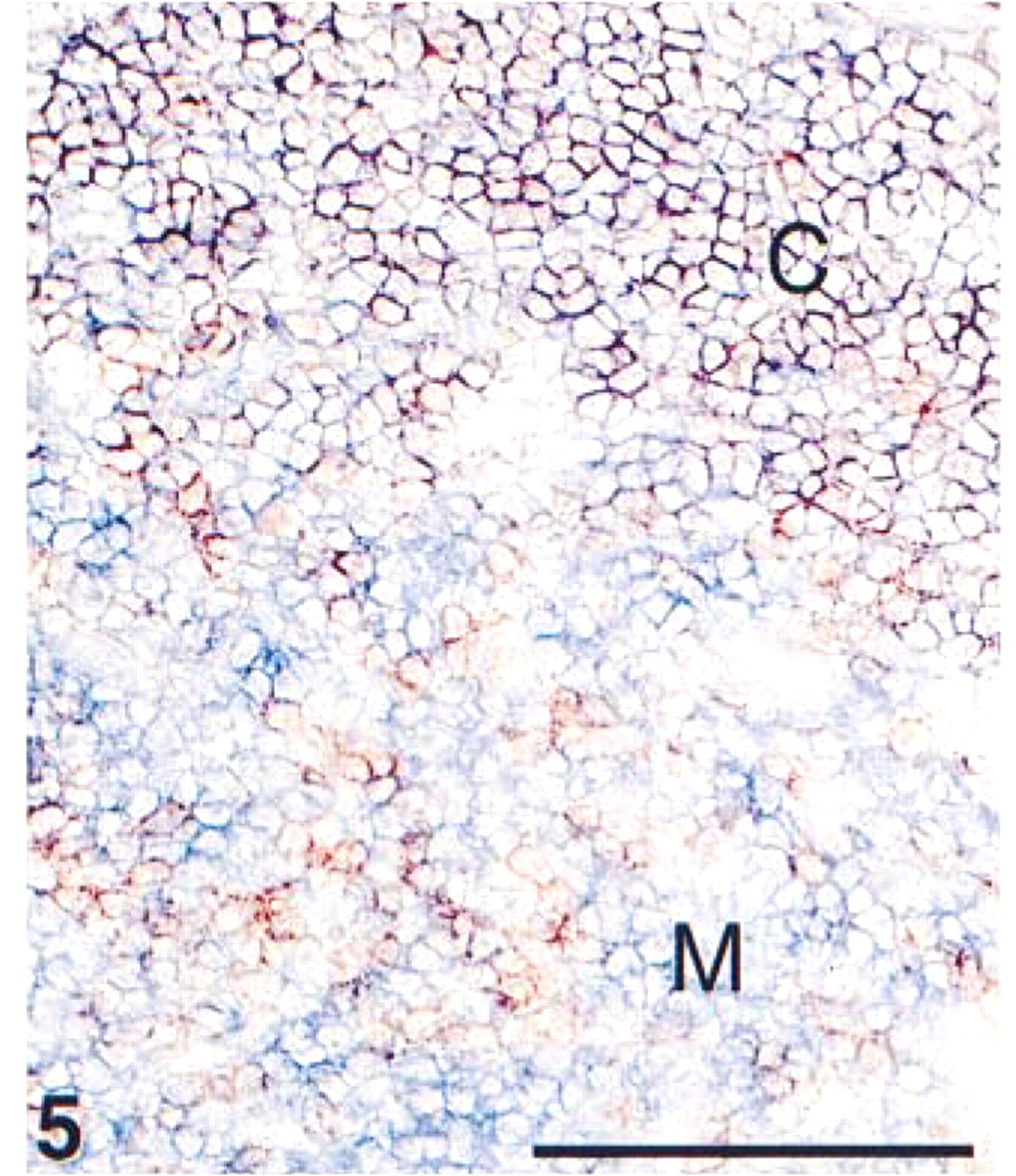
Detail of an acetone-fixed cryostat section from thymus of a 1-year-old infant. Immunohistochemical double staining was performed with ARK-biotinylated anti-CD4 in blue (AP) and anti-CD8 in red (HRP). Immature thymocytes in the cortex (C), co-expressing CD4 and CD8, show a purple mixed color. Matured thymocytes in the medulla (M) stain either single blue (CD4, T-helper/inducer cells) or single red (CD8, T-suppressor/cytotoxic cells). Bar = 100 μm.
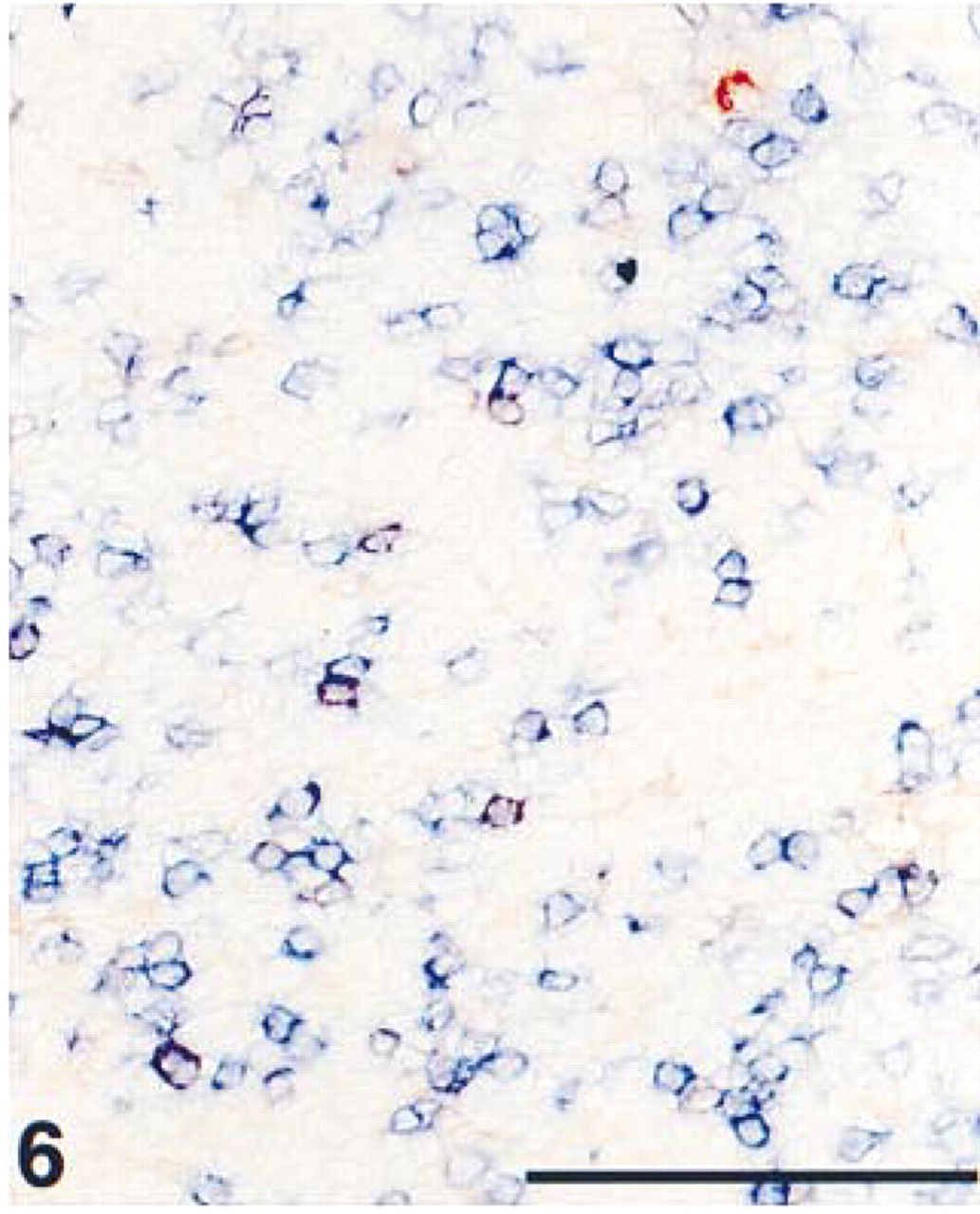
Detail of an acetone-fixed cryostat section from tonsil, showing the parafollicular area. Immunohistochemical double staining was performed with ARK-biotinylated anti-CD3 in blue (AP) and anti-CD25, interleukin-2 receptor in red (HRP). Resting T-lymphocytes stained single blue; activated CD25-positive T-lymphocytes showed a purple mixed color. Some CD25-positive monocytes stained single red. Bar = 100 μm.
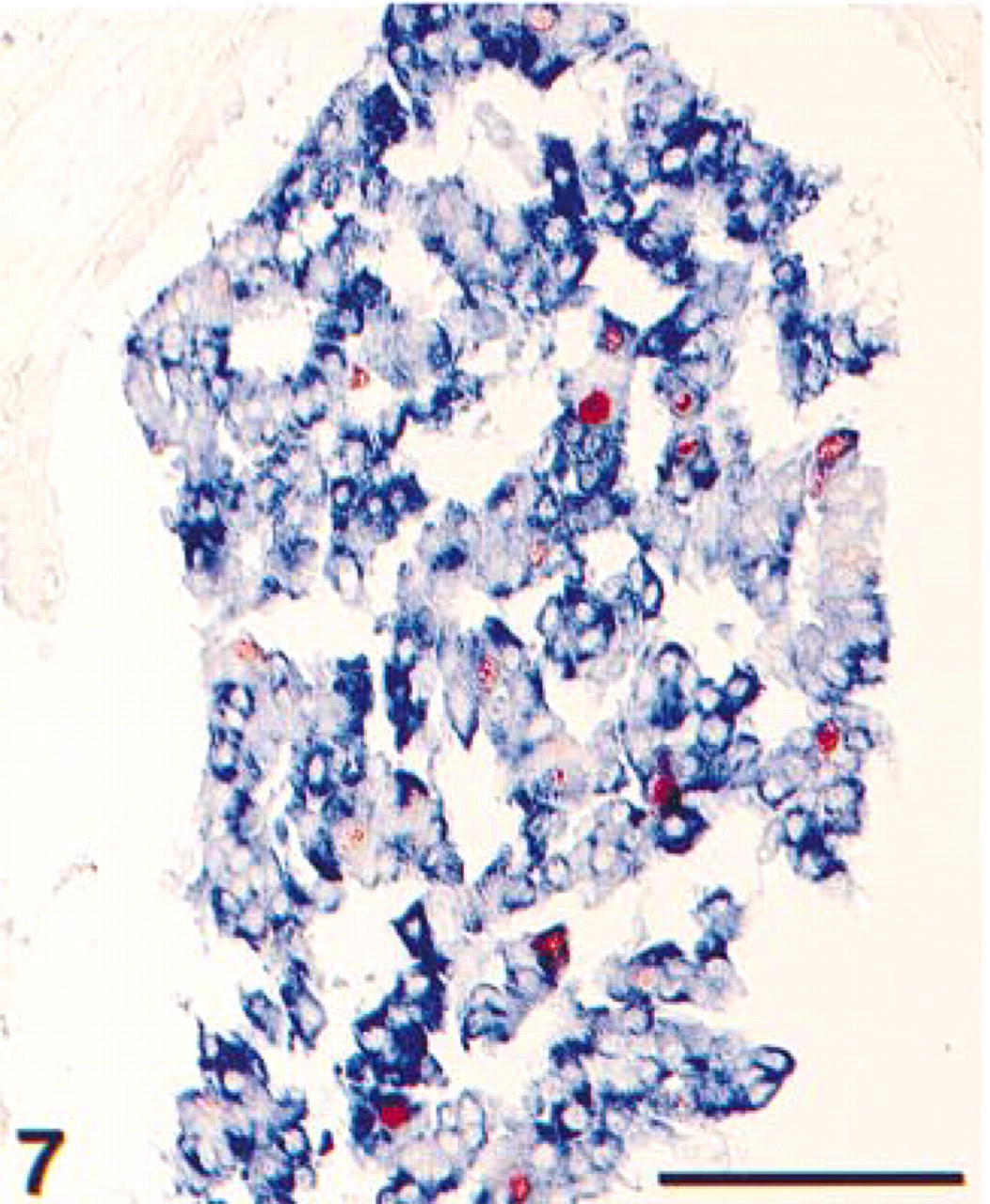
Detail of formalin-fixed, paraffin-embedded breast carcinoma. The tissue section was pretreated with heat-induced antigen retrieval using citrate pH 6.0. Immunohistochemical double staining was performed with ARK-biotinylated anti-cytokeratin, showing a blue (AP) cytoplasmic staining of the epithelial cells and anti-MIB-1 showing proliferating nuclei in red (HRP). Because of separate cell compartments, a purple mixed color is absent. Bar = 100 μm.
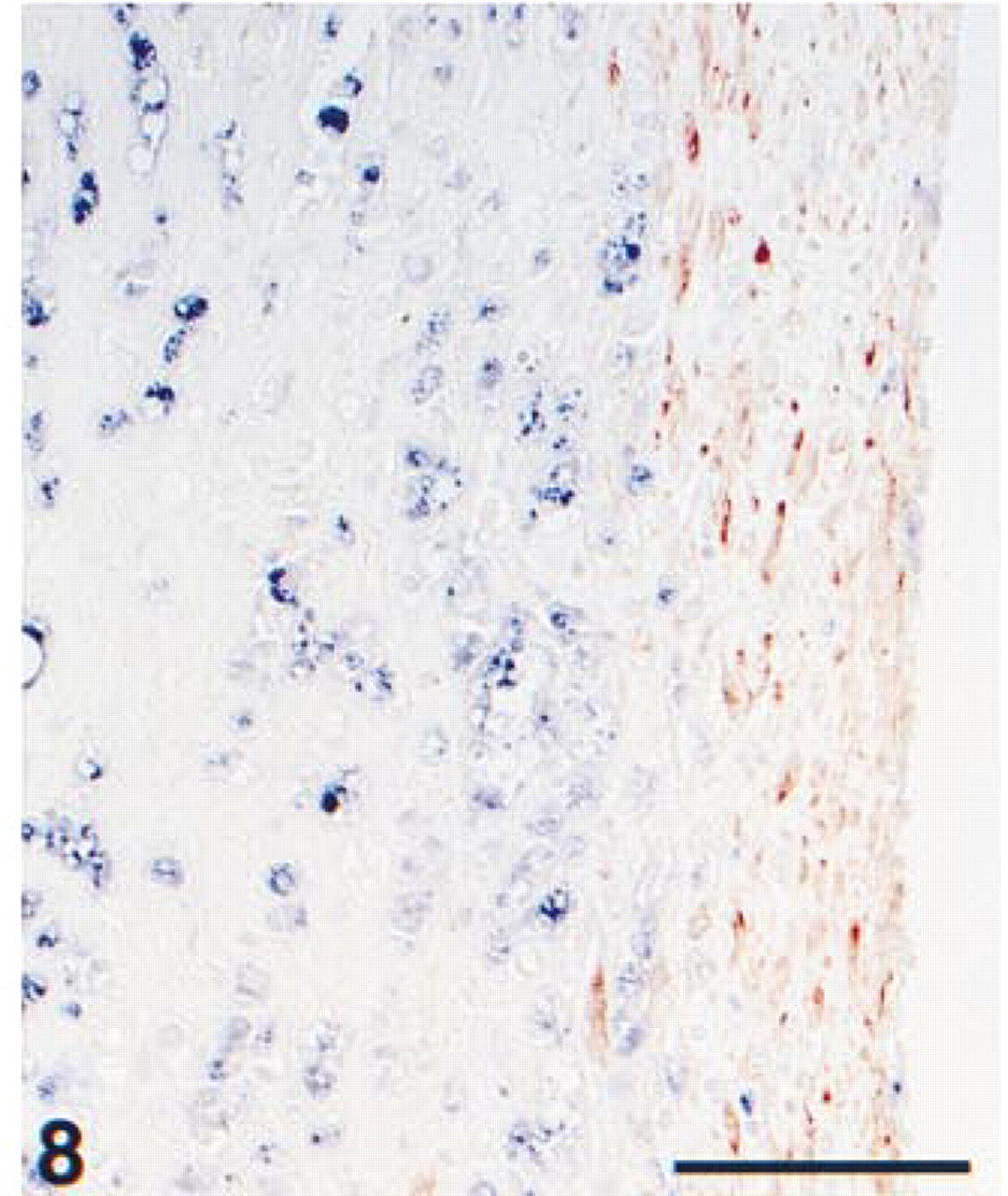
Formalin-fixed, paraffin-embedded carotid artery, showing the thin fibrotic cap at the intima of an atherosclerotic lesion. The tissue section was pretreated with heat-induced antigen retrieval using citrate, pH 6.0. Immunohistochemical double staining was performed with ARK-biotinylated anti-α-actin, staining smooth muscle cells in red (HRP) and anti-CD68, showing macrophages in blue. Because of separate cell types, co-localization is absent. Bar = 100 μm.
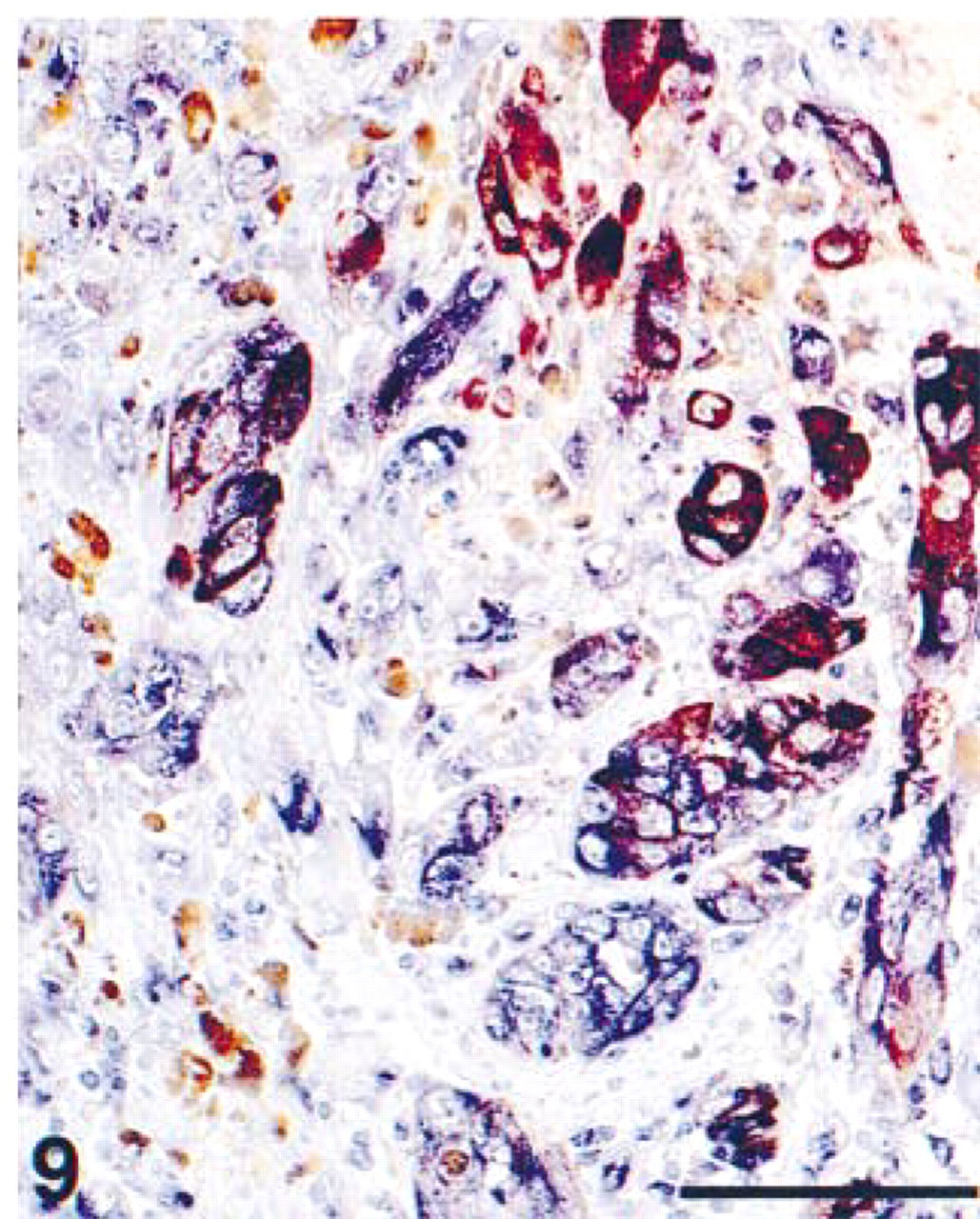
Detail of formalin-fixed, paraffin-embedded skin melanoma. The tissue section was pretreated with heat-induced antigen retrieval using citrate, pH 6.0. Immunohistochemical double staining was performed with ARK-biotinylated anti-HMB45, showing tumor cells in red (HRP), and anti-vimentin, showing all cells of mesenchymal origin in blue (AP). A number of HMB45-positive tumor cells also express vimentin, marked by a purple mixed color at sites of co-localization. Brown melanin pigment is abundantly present. Bar =100 μm.
After application of all the immunoreagent steps, first AP activity was developed in blue (5–15 min, RT) using Tris-HCl buffer (100 mM, pH 8.5) and including naphthol-AS-MX-phosphate as substrate (0.2 mg/ml) and 1 mM levamizole (0.12 mg/ml) as inhibitor of endogenous AP activity. Just before use, Fast Blue BB was added as chromogen (0.2 mg/ml). After rinsing the sections with TBS buffer, HRP activity was developed in red (5–15 min, RT) using AEC as chromogen (0.5 mg/ml) in sodium acetate buffer (50 mM, pH 5.2). Just before use, 0.01% perhydrol was added as substrate.
Controls consisted of replacing one of the primary antibodies with a nonimmune mouse antibody of identical subclass. Specific IgG concentration and Ig isotype/subclass were matched with the specific primary antibody from the original double staining experiment. For the CD3/CD25 combination, the detailed conditions of the matched IgG1 controls are given in Table 1. All primary antibodies in Table 1 were also used for single immunostaining, using the two-step EnVision/HRP technique. After the double staining experiments, a comparison was made with the original single staining slides.
Results
The two multistep double staining experiments, according to the scheme in Figure 1 using either an ARK–biotinylated or an ANHS–biotinylated anti-CD68, EBM11 antibody performed on serial sections showed identical results with respect to both single and double stained structures (Figures 2 and 3). Three additional antibody combinations on cryostat sections are shown in Figures 4–6: von Willebrand factor/E-selectin, CD4/CD8, and CD3/CD25. Figures 7–9 represent double staining experiments performed on paraffin sections after heat-induced antigen retrieval with citrate buffer: Ki67/cytokeratin, vimentin/HMB45, and CD68/α-actin.
All primary antibodies involved in this study displayed comparable staining patterns with either single or double immunoenzyme staining and matched the staining pattern descriptions provided by the supplier. In all performed control experiments, the replaced antibody revealed completely negative staining.
Discussion
In this study we have demonstrated equal double staining results with either ARK-biotinylation or a true biotin conjugate, thus showing that the method of biotinylation did not affect the outcome. Therefore, the DAKO ARK, although designed for different purposes, can be used successfully for in vitro biotinylation of unlabeled anti-human mouse MAbs to be used in a multistep double staining protocol. The obvious advantage of ARK-biotinylation is the requirement of only minute amounts of a primary antibody compared with traditional ANHS-biotin conjugation. Eight tested antibody combinations show distinct basic colors and occasionally a mixed color at sites of co-localization. Unwanted crossreactions among the applied reagents were never observed.
An alternative double staining procedure with two antibodies of similar IgG subclass resembling the present technique, was described by Eichmüller et al. (1996). In our opinion, these authors used a less optimal double staining procedure because of the following reasons, which are overcome in the presently described double staining procedure. (a) The in vitro labeling of a primary antibody with biotinylated whole anti-mouse Ig antibody instead of the presently used biotinylated Fab fragments will result in large complexes, which may lead to avidity problems with the primary antibody. (b) The incubation sequence of alternating immunoreagent steps and enzymatic developments is less ideal compared to the application of all immunoreagents first, followed by two subsequent enzymatic developments. (c) The first staining sequence using a classical two-step detection has rather poor sensitivity/efficiency compared to the presently applied EnVision polymer two-step technique. (c) An accurate calculation of the used biotinylation reagent volume based on the mouse primary antibody Ig concentration is lacking.
Nevertheless, some limitations of the present double staining procedure should be noted. First, the ARK-biotinylation is based on a calculation requiring a specific mouse Ig concentration. Consequently, only purified commercial antibody products with a known mouse immunoglobulin concentration, which should be stated in the data sheet, can be properly employed for in vitro ARK-biotinylation. Second, the observation of a mixed color at sites of co-localization is generally limited to two antigens that occur with almost equal staining intensities (Van der Loos 1999). When the antigens are present at varying antigen densities, one colored reaction product may overwhelm the other. In such instances, separate observations applying a two-color double fluorescence technique (Brandtzaeg et al. 1997) or a combination of a fluorescent AP technique with epipolarization of an immunogold/silver method (Van der Loos and Becker 1994) might be preferred. Finally, the blue AP reaction product using Fast Blue as chromogen is rather diffusely localized and is not as sensitive/efficient compared with HRP activity visualization using aminoethylcarbazole. Therefore, the application of AP in blue is restricted to strongly expressing antigens independent of a sharp localization. The present double staining protocol offers the possibility of a reversed color combination, either using differently labeled EnVision and streptavidin reagents or selecting the other primary antibody for ARK–biotinylation (Table 2).
Although the eight tested double staining combinations in this study still are limited, one can assume that the ARK–biotinylation procedure, in combination with the multistep indirect/direct double staining technique, can be theoretically applied for an indefinite number of mouse monoclonal primary antibody pairs. This double staining protocol is completely independent of the mouse antibody Ig (sub)class and the availability of primary antibodies in a conjugate format. The general protocol outlined in Table 2 may serve as a “blueprint” for many successful double staining combinations.
Footnotes
Acknowledgements
We wish to thank DAKO A/S (Glostrup, Denmark) for financial support for color print reproduction and Mr Mikkel Nielsen for supporting us with reagents and information related to the ARK. We are also indebted to Prof Dr Anton E. Becker for critical reading of our manuscript.