
Abstract
Acidic fibroblast growth factor (aFGF) is a heparin binding protein that displays pleiotropic activity. The purpose of this study was to document the presence of the translated aFGF product, its mRNA, and its location in the colon. mRNA was extracted from bovine large intestine and reverse transcribed to cDNA. Nested-primer PCR was used to determine the presence of mRNA using primers homologous to the previously published bovine aFGF cDNA. Purification of translated aFGF was performed using an established HPLC protocol. Western blot analysis of the HPLC fractions was performed using two epitopeindependent antibodies against aFGF. Immunohistochemistry employed these antibodies to determine the locus of aFGF expression. The nested-primer PCR product of predicted size was homologous to the published bovine aFGF mRNA sequence, as determined by DNA sequencing. Intestinal aFGF had a mass similar to bovine aFGF isolated from other tissues, and immunocrossreacted with two peptide-based, epitope-independent anti-aFGF antisera on Western blotting. Immunohistochemical analysis of large intestine using these two independent antisera localized aFGF within the myenteric plexus. These data demonstrate that aFGF is present within the myenteric plexus of the enteric nervous system.
A
aFGF has been shown to be strongly associated with the neuronal and support cells of the central and peripheral nervous systems (CNS, PNS). The adult brain, from which aFGF was originally purified, is one of the most abundant sources of the protein (Thomas et al. 1984, 1991). aFGF has been shown by immunohistochemistry to be localized within specific subsets of neurons in regions associated with motor and sensory functions (Eckenstein et al. 1991; Fallon et al. 1992). Purified aFGF is mitogenic for brain neuroblasts and promotes neurite extension from spinal cord neurons (Wu et al. 1988; Iwasahi et al. 1995). In addition, aFGF induces expression of nerve growth factor and is mitogenic for astrocytes in culture (Pettmann et al. 1985; Yoshida and Gage 1991). Endogenous aFGF appears to be particularly abundant in sciatic nerve of the PNS and decreases after neuronal transection (Elde et al. 1991). Application of human recombinant aFGF (raFGF) was shown to promote motor nerve regeneration as measured by electrophysiological analysis (Walter et al. 1993). These data suggest a mitogenic role for aFGF in the CNS and PNS.
Associated with the PNS is the enteric nervous system (ENS). The ENS is composed of the outer myenteric plexus, located between the longitudinal and circular muscle layers of the muscularis externa, and the inner submucosal plexus, situated at the interface of the circular muscle layer and the submucosa. These ganglia are composed of neurons and astrocyte-like glial (Gershon 1981; Gershon and Rithman 1991). We report the presence of aFGF mRNA and protein in the bovine large intestine. We also show by immunohistochemistry with two peptide-based, epitope-independent antibodies that aFGF is localized within the myenteric plexus of the ENS.
Materials and Methods
Preparation of Bovine Large Intestine
Large intestine was prepared from healthy animals at Branchberg Farms (Merck; Branchberg, NJ). All procedures for preparation of the organs were performed at this abatoire. Animals were quickly sacrificed by severing the carotid arteries and jugular veins. Large intestine was removed from the slaughtered animal immediately. The organ was cut along the longitudinal axis, its contents removed, and the mucosa washed clean of all debris with ice-cold PBS, pH 7.4. The organ was then cut into strips of 1-3 cm in width and 8-10 cm in length. For the purposes of purification of aFGF, intact strips were bagged, sealed, and immersed in wet ice for transport to the laboratory where the extraction procedure was performed (see below). For immunohistochemical studies, the strips were trimmed into 0.5-cm2 segments. These segments of bovine large intestine were fixed with 4% zinc formalin, rinsed with double-distilled water, placed in optimal cutting temperature fluid, and snap-frozen in liquid N2 at the abatoire for transport to the laboratory for immunostaining (see below). The turnaround time for this preparation was limited to 5 min by processing only a small portion of the large intestine. Hematoxylineosin staining of the frozen sections of large intestine showed preservation of the histological architecture, with intact epithelial and goblet cells comprising the crypts of Lieberkuhn. The discrete regions of the muscularis mucosa, lamina propria, submucosa, and muscularis externa were also observed as compared to histology textbooks. Furthermore, the circular and longitudinal muscle layers of the muscularis externa were clearly observed.
DNA Cloning and Sequencing
Total RNA was obtained from 0.8-1.0-g sections of bovine large intestine snap-frozen in liquid N2 using a protocol described for the Total RNagents kit (Promega; Madison, WI). Five μg of total RNA was annealed to oligo(dT) primers and the mRNA was reverse transcribed to cDNA at 42C for 50 min (Superscript Amplification kit; Gibco BRL, Grand Island, NY). Polymerase chain reaction (PCR) amplifications were performed using AmpliTaq DNA polymerase (Perkin Elmer; Norwalk, CT). Sense 5′ CTCCTCTACTGCAGC-AAC 3′ (999-1016) and antisense 5′ AGATTTCTTTAAT-CAGAGGAGACTG 3′ (1369-1393) primers, based on the published bovine-specific aFGF sequence (Halley et al. 1988), were used to amplify a 395-bp product through 40 cycles (1 min at 94C, 2 min at 58C, 2 min at 72C), followed by a single 10-min extension at 72C. The PCR product was submitted to a second round of amplification using nested sense 5′ CCAGATGGCACAGTGGATG 3′ (1041-1059) and antisense 5′ AGTGAGTCCGAGGACCG 3′ (1319-1335) primers (30 cycles of 1 min at 94C, 2 min at 58C, 2 min at 72C) to generate a 295-bp product. The third PCR amplification was performed with nested sense 5′ CAGCT-GCAGCTCTGTGC 3′ (1089-1105) and antisense 5′ CCAATGCTTCTCTGCATG 3′ (1266-1283) primers that generated a 195-bp product. PCR experiments were performed in parallel with actin provided by Gibco BRL as a control. Bovine large intestine total RNA not reverse transcribed to DNA showed no PCR signals, thus eliminating the possibility that the amplified DNA was the result of contamination by genomic DNA.
The PCR products were gel-purified (Wizard PCR DNA kit; Promega), ligated into the pCRII vector by TA cloning (TA Cloning kit; Invitrogen, San Diego, CA), and transformed into competent E. coli (One Shot; Invitrogen). The DNA was gel-purified (DNA Wizard Miniprep; Promega) and sequenced by the chain-termination method using T7 DNA polymerase (Sanger et al. 1977) (Sequenase Version 2.0; United States Biochemical, Cleveland, OH).
Synthesis of Human Recombinant aFGF
Human recombinant aFGF (raFGF) was obtained by expression in E. coli of the gene encoding for the 140-amino-acid form of aFGF and purified to homogeneity using previous methods (Linemeyer et al. 1987). raFGF was used as an ∼16-kD molecular weight standard in SDS-PAGE and Western blots.
Purification of aFGF
The strips of ice-cold bovine large intestine obtained at the abatoire were homogenized in a Waring blender at 4C in the presence of a mixture of protease inhibitors (1 mM each of benzamidine, EDTA, phenylmethylsulfonyl fluoride, and N-tosyl-L-phenylalanine chloromethyl ketone), centrifuged, and precipitated by (NH4)2SO4 fractionation from 1.5 to 3.4 M as described (Thomas et al. 1984). Unless otherwise noted, subsequent purification steps were also done at 4C. The centrifuged pellet was resuspended in 100 mM sodium phosphate, pH 6.0, batch-adsorbed to CM Sephadex C-50 (Pharmacia; Stockholm, Sweden) equilibrated with 150 mM NaCl, 100 mM sodium phosphate, pH 6.0, washed to removed unadsorbed proteins, and eluted with 0.6 M NaCl in phosphate buffer. Peak fractions were pooled and diluted with 10 mM sodium phosphate, pH 7.2, until the conductivity was <12 mS and batch-adsorbed to heparin-Sepharose CL-6B (Pharmacia) for 1-2 hr with gentle stirring. The resin was rinsed with 0.8 M NaCl, 10 mM sodium phosphate, pH 7.2, until the absorbance reached baseline and bound protein was eluted with 1.2 M NaCl and injected onto a 5 × 0.46 cm C4 reversed-phase HPLC column (Vydac; Western Analytical Products, Murietta, CA) equilibrated with 10 mM trifluoroacetic acid (TFA). Proteins were fractionated with a 30-min linear gradient of 0-66% acetonitrile in 10 mM TFA at a flow rate of 0.5 ml/min at 21C. Samples containing ∼16 kD immunocrossreactive aFGF on Western blots of SDS polyacrylamide electrophoretic gels were rechromatographed on a 5 × 0.46 cm microbore C4 reversed-phase column (Vydac) equilibrated in 10 mM TFA and eluted with a 90-min linear gradient of 25-50% acetonitrile at 25 μl/min controlled by a Pharmacia LKB SMART system. Aliquots of fractions were analyzed for heparin-dependent growth activity (Thomas 1993) using the Balb/c 3T3 fibroblast (ATCC; Manassas, VA) serum-free mitogenic assay as previously described (Ortega et al. 1991). The microbore peak that showed heparin-dependent mitogenesis with an ED50 similar to that of raFGF (Ortega et al. 1991; Thomas 1993) was analyzed for size and the presence of aFGF using SDS-PAGE and Western blot analysis, respectively.
SDS-PAGE and Western Blotting
Samples were fractionated by 15% SDS-PAGE. The proteins in the gel were precipitated in 50% trichloroacetic acid, equilibrated in 50% EtOH, 7.5% HOAc, and fixed with 10% glutaraldehyde for 15 min. Gels were washed with multiple water changes for 1-2 days and protein bands were silver-stained. For Western blots, SDS-PAGE-fractionated proteins were electrophoretically transferred to ProBlott membranes (Collaborative Biomedical Research; Waltham, MA). The proteins were crosslinked to the membrane with 0.5% glutaraldehyde in PBS (v/v) to increase the retention of proteins and thereby enhance the sensitivity of the Western blot (Karey and Sirbasku 1989). Excess crosslinking activity was then blocked with 4% ethanolamine in PBS (v/v), pH 7.4. Nonspecific absorption sites were blocked with 0.25% gelatin (Bio-Rad Laboratories; Hercules, CA), 0.05% Tween-20 (Bio-Rad) for 2 hr. Epitope-independent antisera generated to human raFGF amino acid sequences 23-33 (LRILPD-GTVDG) and 128-140 (KAILFLPLPVSSD) (Fallon et al. 1992) were used in the range of 1:1000-5000 and 1:5000-30,000 dilution, respectively. These amino acid sequences show 100% homology between human and cow (Fallon et al. 1992). Control studies included performing the Western blots with preimmune serum and with raFGF-adsorbed anti-aFGF antibodies. Bound antibodies were complexed with [125I]-protein A (Amersham; Princeton, NJ) and autoradiographically visualized as described (Fallon et al. 1992).
Immunohistochemistry
Formalin-fixed, snap-frozen colon samples were cut with a cryostat into 4-μm-thick sections, postfixed with Bouin's solution (4% formaldehyde, 0.4% picric acid, 100 mM HOAc), and washed with PBS, pH 7.4 Cells were permeabilized with 0.5% Triton X-100 in PBS for 15 min at 21C. Endogenous peroxidases were quenched with 3% H2O2 in 70% methanol for 15 min and nonspecific sites were blocked with goat serum (BioGenex; San Ramon, CA) for 30-60 min. Epitope-independent antisera to amino acid residues 23-33 and 128-140 of aFGF were diluted in 1.5% globulin-free bovine serum albumin (BSA) (Sigma; St Louis, MO) in PBS at 1:25-50 and 1:100-500 dilutions, respectively, for 1 hr at 21C. Polyclonal rabbit antiserum to neuron-specific enolase (NSE) (BioGenex), used to document the presence of neuronal cells, was added to the sections for 30 min, using the manufacturer's dilution as provided with the antibody (Scheuermann et al. 1989). Sections were sequentially incubated with biotinylated goat anti-rabbit IgG H+L (Vector Laboratories; Burlingame, CA) diluted 1:200 with 1.5% BSA in PBS for 30 min, peroxidase-labeled avidin-biotin (Vector Elite ABC reagent) in 150 mM NaCl plus 10 mM sodium phosphate, pH 7.4, or streptavidin (BioGenex) in 1% albumin in PBS for 30 min, and 0.15% diaminobenzidine (DAB) in cacodylate buffer, pH 6.0, plus 0.04% H2O2 for 30 min in the dark. Control studies included preimmune sera at the identical dilutions for the primary antibodies indicated above, with the secondary antibody alone, and in the presence of the primary antibodies preadsorbed with raFGF. Sections were counterstained with hematoxylin and eosin, dehydrated with 50, 70, 95, and 100% ethanol sequentially, and mounted with a coverslip and Permount-sealed. DAB signal over background in immunostained sections using the peroxidase-labeled avidin-biotin detection system with DAB color development was optimal using the double-fixation method of 4% zinc formalin followed by Bouin's solution, relative to formalin alone or Bouin's solution alone.
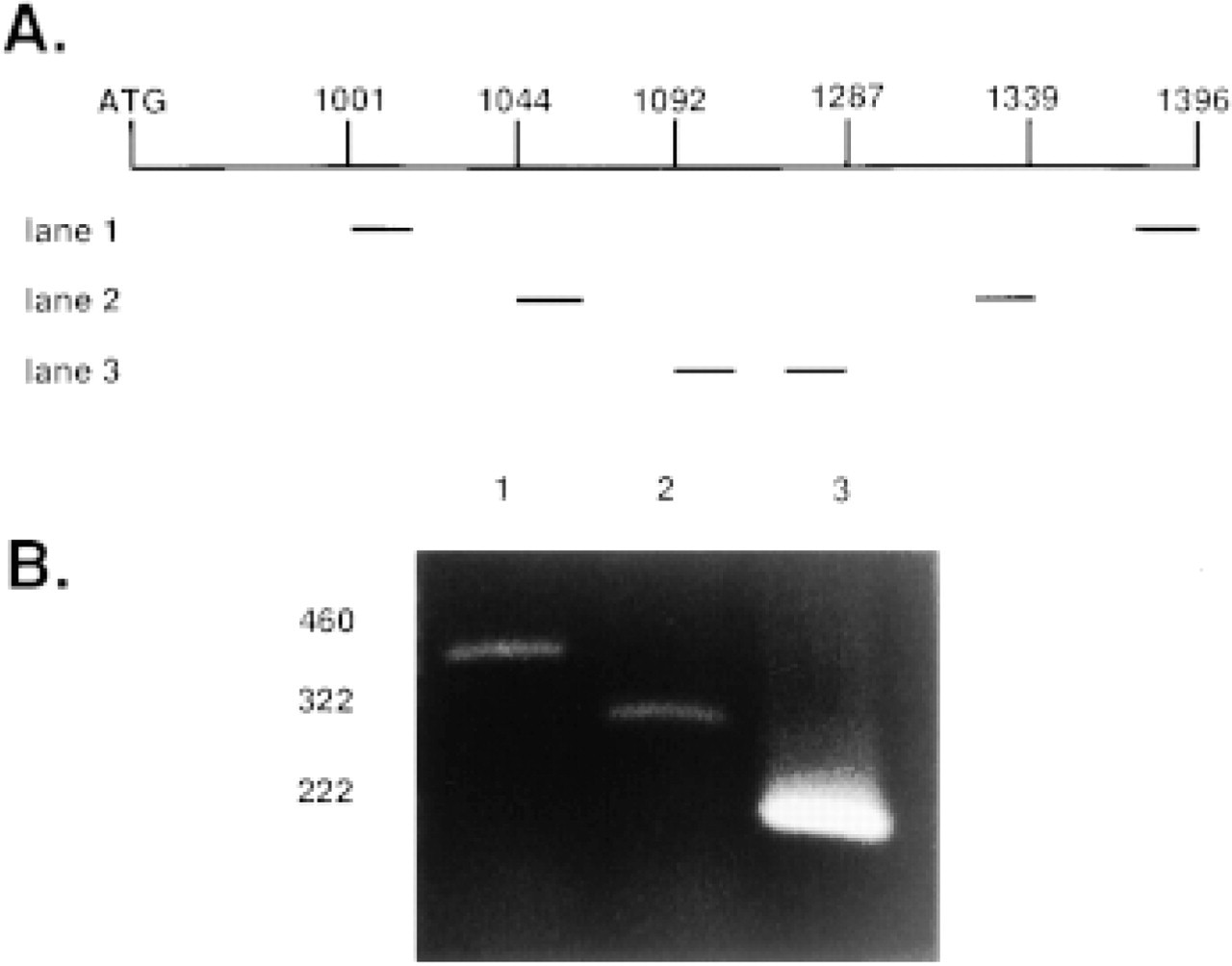
Detection of aFGF mRNA in bovine large intestine. (
Results
Acidic FGF mRNA Is Present in Bovine Intestine
In general, mRNAs encoding highly active growth factors are present in low abundance. Therefore, RT-PCR was used to test for the presence of aFGF mRNA in the bovine large intestine. This first set of bovine aFGF-derived primers (Figure 1A, Lane 1) amplified a 395-bp PCR product (Figure 1B, Lane 1). To increase sensitivity and specificity, the PCR cDNA product was used as a template for a second amplification using primers nested within the first PCR product (Figure 1A, Lane 2), which resulted in the predicted 295-bp band (Figure 1B, Lane 2). A final PCR amplification using primers nested within the second PCR product (Figure 1A, Lane 3) resulted in an intense band of the predicted 195-bp size (Figure 1B, Lane 3), further confirming the identity of the aFGF mRNA. The DNA sequence of this 195-bp PCR product matches that of the published bovine aFGF cDNA sequence, which differs from the published human aFGF sequence by 10-bp in this region (Jaye et al. 1986; Halley et al. 1988).
Purification of aFGF from Bovine Large Intestine
We sought to confirm the presence of aFGF in bovine large intestine by purification using methods similar to those previously employed to isolate it from other sources (Thomas et al. 1984; Gimenez-Gallego et al. 1986). Supernatants of crude homogenates were fractionated by differential (NH4)2SO4 precipitation and sequential cation exchange, heparin affinity, and reversed-phase HPLC chromatography (see Materials and Methods). Balb/c 3T3 mitogenic activity was eluted by conditions characteristically used to recover aFGF and was potentiated by 50 μ/ml of heparin, also typical of aFGF (not shown) (Thomas et al. 1984).
To purify bovine intestinal aFGF to apparent homogeneity, the active fraction was re-chromatographed on a microbore (5
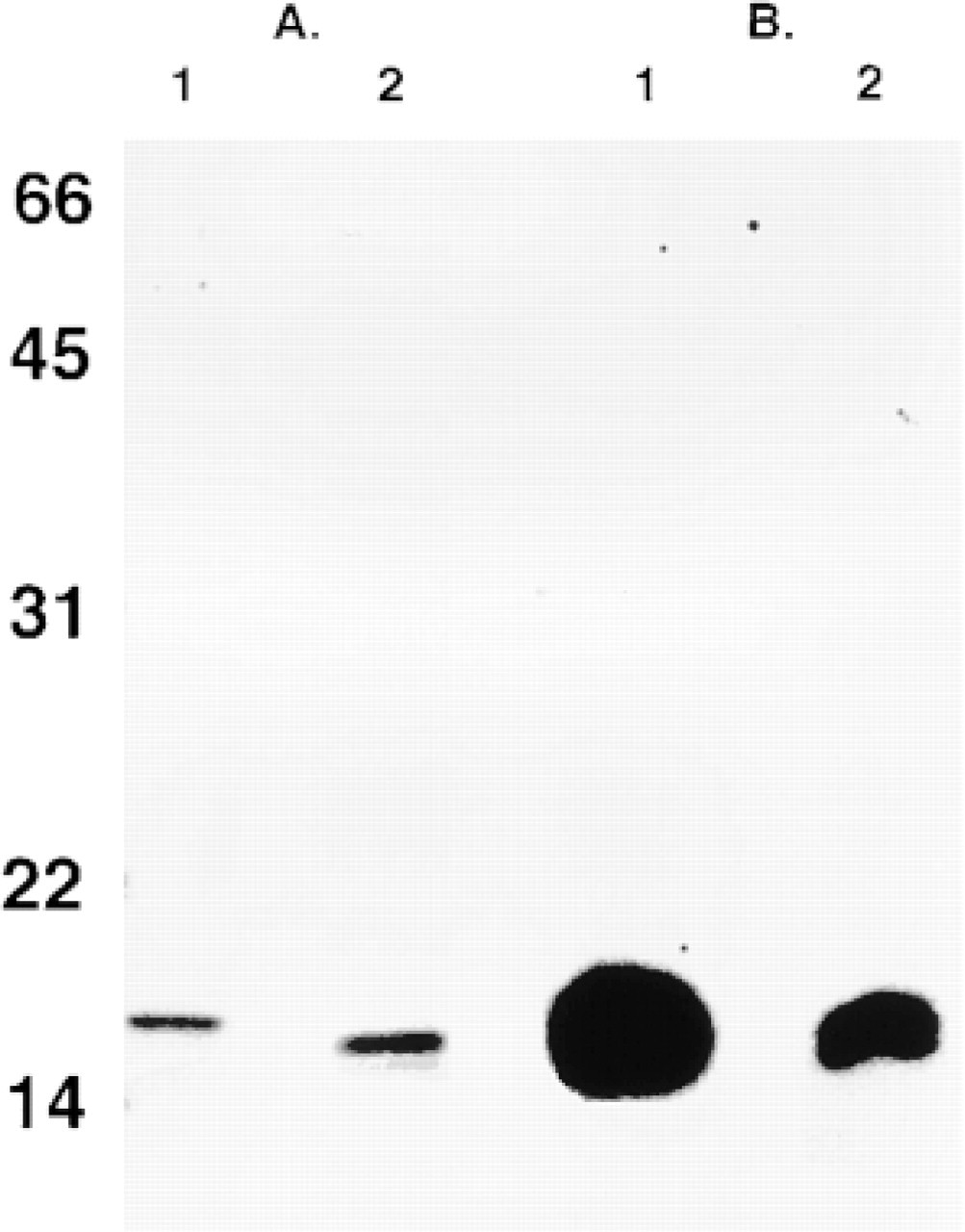
Purity and immunocrossreactivity of aFGF purified from bovine large intestine. Samples were fractionated by electrophoresis in 15% SDS-PAGE, followed by silver staining for observation of the protein masses. Masses (kD) of standard proteins are denoted at left. (
Immunohistochemistry
To determine the location of aFGF in bovine large intestine, 4-μm-thick frozen-fixed sections were analyzed with the two epitope-independent anti-aFGF antisera. Strong immunocrossreactivity of cells residing between the longitudinal and circular muscle layers comprising the muscularis externa were observed with C-terminal (Figure 3B) and N-terminal (Figure 3D) peptide-based anti-aFGF antisera. Sections treated with the corresponding C- and N-terminal preimmune sera (Figures 3A and 3C, respectively) did not display staining. Similarly, neither peptide-based antisera adsorbed with recombinant human aFGF or secondary antibody alone exhibited significant signal over background in the myenteric plexus (not shown). Anti-aFGF antisera-stained cells were shown to co-localize with neuronal ganglia by immunohistochemistry with an antiserum that recognized the γ-subunit of NSE (Figure 3F). Immunohistochemistry performed with the secondary antibody without the NSE primary antiserum showed no signal (Figure 3E). Cell bodies in the mucosa, muscularis mucosa, submucosa, and muscularis externa did not stain with either the N- or the C-terminal-specific epitope-independent anti-aFGF antibody. Anti-aFGF staining is therefore localized within the myenteric plexus of the ENS.
Discussion
Bovine aFGF has previously been found in subsets of neurons within the central and peripheral nervous systems. The large, well-organized ENS that innervates the intestine contains an extensive network of ganglia that control muscle contractions required for the propulsion of enteric content. Therefore, given the presence of aFGF in both the CNS and the PNS, and the size and importance of the ENS, we examined intestinal tissues for the presence of aFGF.
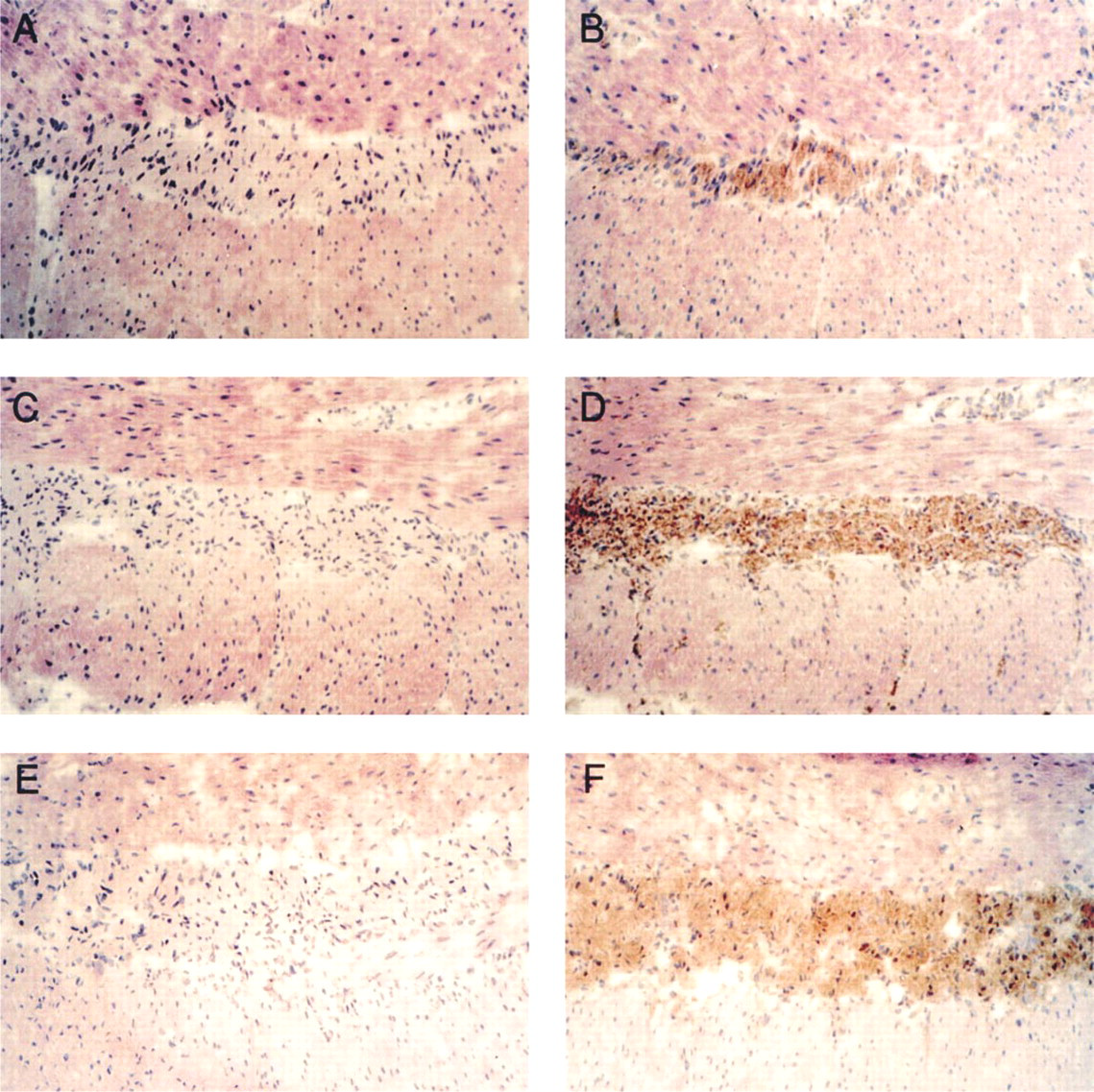
Immunolocalization of aFGF in bovine large intestine. Immunohistochemistry was done using 4-μm-thick frozen sections of bovine large intestine. (
RNA from bovine large intestine was found to contain aFGF mRNA established by nested-primer PCR amplification and confirmed by DNA sequence analysis. However, because of differences in translational efficiency among mRNAs and different relative stabilities of mRNA and protein, the presence of mRNAs does not always reliably indicate the abundance of the corresponding proteins. For example, human aFGF mRNA contains a stable 5′ hairpin loop that probably inhibits translational initiation, so it need not be indicative of the presence of biologically significant amounts of the protein product (Forough et al. 1991).
To confirm the presence of aFGF translated product in extracts of bovine large intestine, we monitored HPLC fractions for heparin-dependent mitogenic activity (a characteristic of aFGF but not of bFGF) and confirmed the purity of the mitogenic fraction with SDS-PAGE in parallel with Western blot analysis. Bovine intestinal aFGF bound and eluted from ion exchange, heparin-Sepharose, and reversed-phase HPLC columns under conditions characteristic of aFGF purified from other sources. Silver-stained SDS-PAGE of the heparin-dependent mitogenic fraction revealed a closely spaced doublet of 15-16 kD characteristic of partial N-terminal proteolysis of aFGF purified from bovine brain (Thomas et al. 1984; Gimenez-Gallego et al. 1986; Thomas et al. 1993). Two peptide-based, epitope-independent peptide antisera against amino acid sequences near the amino terminal (23-33) and at the carboxy terminal (128-140) of aFGF revealed immunocrossreactivity at the same ∼16-kD mass. The peptides used to generate the two antibodies have limited homology between aFGF and the other FGF family members (Fallon et al. 1992). In addition, a 100-and 500-fold increase in bFGF is required to observe Western blot signals using the anti-(23-33)- and anti-(128-140)-aFGF, respectively. In all samples of bovine large intestine that exhibited heparin-dependent mitogenic activity, Western blot analysis showed signal with both antibodies, indicating specificity for aFGF. A signal observed with only one antibody or no observed signal would have indicated nonspecificity. Therefore, using standard techniques for the purification of aFGF, a product of appropriate mass, bioactivity, and specific immunocrossreactivity for aFGF was isolated.
Immunohistochemistry with these same two peptide-based, epitope-independent antisera showed that aFGF was localized within the myenteric plexus of the ENS. The location of the myenteric plexus was confirmed by NSE immunoreactivity (Figures 3E and 3F). This NSE immunoreactivity was not present in the surrounding circular and longitudinal smooth muscle cell layers. The immunohistochemical staining was specific by several criteria, including the equivalent staining by two epitope-independent anti-aFGF antisera (Figures 3B and 3D) and the absence of staining with preimmune sera (Figures 3A and 3D), immune sera preadsorbed with aFGF, and secondary antisera alone (not shown). Although either epitope-independent anti-aFGF antiserum might recognized unrelated proteins with randomly similar epitopes, the co-distribution of such nonspecific immunocrossreactivity would be very unlikely. Therefore, the use of two peptide-based, epitope-independent antisera, previously used to map the distribution of aFGF within brain, virtually eliminates the risk of incorrect identification of aFGF (Fallon et al. 1992).
While myenteric plexus-derived neurons have been reported to innervate the mucosa (Messenger and Furness 1990; Furness et al. 1991), no overt ramifications from the plexus into the muscle layers were consistently observed with the epitope-independent anti-aFGF antibodies relative to background. No discrete immunostaining was observed in the muscularis mucosa or at the mucosal epithelial cells comprising the crypts of Lieberkuhn in any of the frozen sections of the bovine large intestine. The absence of signal in the epithelial cells indicates, in contrast to previous reports (Hughes and Hall 1993; Yamamoto et al. 1995; el-Hariry et al. 1997), the lack of detectable aFGF expression. These findings could be due to differences in species, methods of tissue fixation, or to nonspecific immunocrossreactivity of the single polyclonal antibody employed by other workers (Hanneken and Baird 1992).
In summary, our data document the presence of aFGF in the myenteric plexus of the large intestine using three independent techniques: presence of mRNA sequence homologous to the published aFGF sequence documented by DNA sequencing of the nested-primer RT-PCR DNA product; immunocrossreactivity of two peptide-based, epitope-independent antibodies employed in Western blot analysis of the final heparin-dependent mitogenic HPLC fraction; and immunohistochemistry of frozen-sectioned colon using the same antibodies. Further biological and genetic studies of intestinal aFGF and its receptors will be required to define the role of this potent mitogenic and neurotrophic factor in gastrointestinal neurobiology, physiology, and pathology.
Footnotes
Acknowledgment
AC thanks the Merck Research Laboratories for postdoctoral fellowship support.