
Abstract
Amelogenin plays a crucial role in tooth enamel formation, and mutations on X-chromosomal amelogenin cause X-linked amelogenesis imperfecta (AI). Amelogenin pre–messenger RNA (mRNA) is highly alternatively spliced, and during alternative splicing, exon4 is mostly skipped, leading to the formation of a microRNA (miR-exon4) that has been suggested to function in enamel and bone formation. While delivering the functional variation of amelogenin proteins, alternative splicing of exon4 is the decisive first step to producing miR-exon4. However, the factors that regulate the splicing of exon4 are not well understood. This study aimed to investigate the association between known mutations in exon4 and exon5 of X chromosome amelogenin that causes X-linked AI, the splicing of exon4, and miR-exon4 formation. Our results showed mutations in exon4 and exon5 of the amelogenin gene, including c.120T>C, c.152C>T, c.155C>G, and c.155delC, significantly affected the splicing of exon4 and subsequent miR-exon4 production. Using an amelogenin minigene transfected in HEK-293 cells, we observed increased inclusion of exon4 in amelogenin mRNA and reduced miR-exon4 production with these mutations. In silico analysis predicted that Ser/Arg-rich RNA splicing factor (SRSF) 2 and SRSF5 were the regulatory factors for exon4 and exon5 splicing, respectively. Electrophoretic mobility shift assay confirmed that SRSF2 binds to exon4 and SRSF5 binds to exon5, and mutations in each exon can alter SRSF binding. Transfection of the amelogenin minigene to LS8 ameloblastic cells suppressed expression of the known miR-exon4 direct targets, Nfia and Prkch, related to multiple pathways. Given the mutations on the minigene, the expression of Prkch has been significantly upregulated with c.155C>G and c.155delC mutations. Together, we confirmed that exon4 splicing is critical for miR-exon4 production, and mutations causing X-linked AI in exon4 and exon5 significantly affect exon4 splicing and following miR-exon4 production. The change in miR-exon4 would be an additional etiology of enamel defects seen in some X-linked AI.
Keywords
Introduction
Amelogenins are highly conserved across species (Salido et al. 1992) and essential for enamel formation to form the main structural matrix during enamel formation. In humans, the amelogenin gene is located on both X and Y chromosomes, but the majority (over 90%) of transcripts are from AMELX (Salido et al. 1992), and in mice, it locates only on the X chromosome as Amel. Both AMELX and Amel pre–messenger RNA (mRNA) are highly alternatively spliced (Salido et al. 1992; Simmer et al. 1994), producing 2 major mRNAs for a matrix protein (M180 in mice and H174 in humans) (Li et al. 2008) and a signaling protein (LRAP) (Gibson et al. 2009). During this event, exon4 is mostly spliced out/skipped. We previously found that a novel miRNA derives from the spliced-out exon4 (miR-exon4) (Le et al. 2016).
Mature microRNAs (miRNAs) are a class of naturally occurring and small noncoding single-stranded RNA consisting of 18 to 24 nucleotides. They bind to target mRNAs to inhibit protein translation and induce mRNA degradation in many organs/tissues, including teeth (Michon et al. 2010), resulting in altered development, apoptosis, and cell cycle regulation (Bartel 2004). Mammalian miRNA loci reside in introns or exons of their pre-mRNA host genes, sharing promoters, or as separate genes transcribed from their promoters (Kim et al. 2009). Among miRNAs, an exonic miRNA is created by the host gene’s alternative splicing or transcription by an independent promoter (Marsico et al. 2013). Thus, alternative splicing of amelogenin exon4 is a fundamental step for miR-exon4 production.
Alternative splicing is regulated by Ser/Arg-rich RNA splicing factors (SRSFs). SRSFs bind to pre-mRNAs at exon splicing enhancer (ESE) sequences to define exons to be included and introns to be skipped (Long and Caceres 2009). Including or skipping an alternative exon during alternative splicing depends on the preferential SRSFs binding on the alternative or conserved exons (Keren et al. 2010; Han et al. 2011) (Fig. 1A).
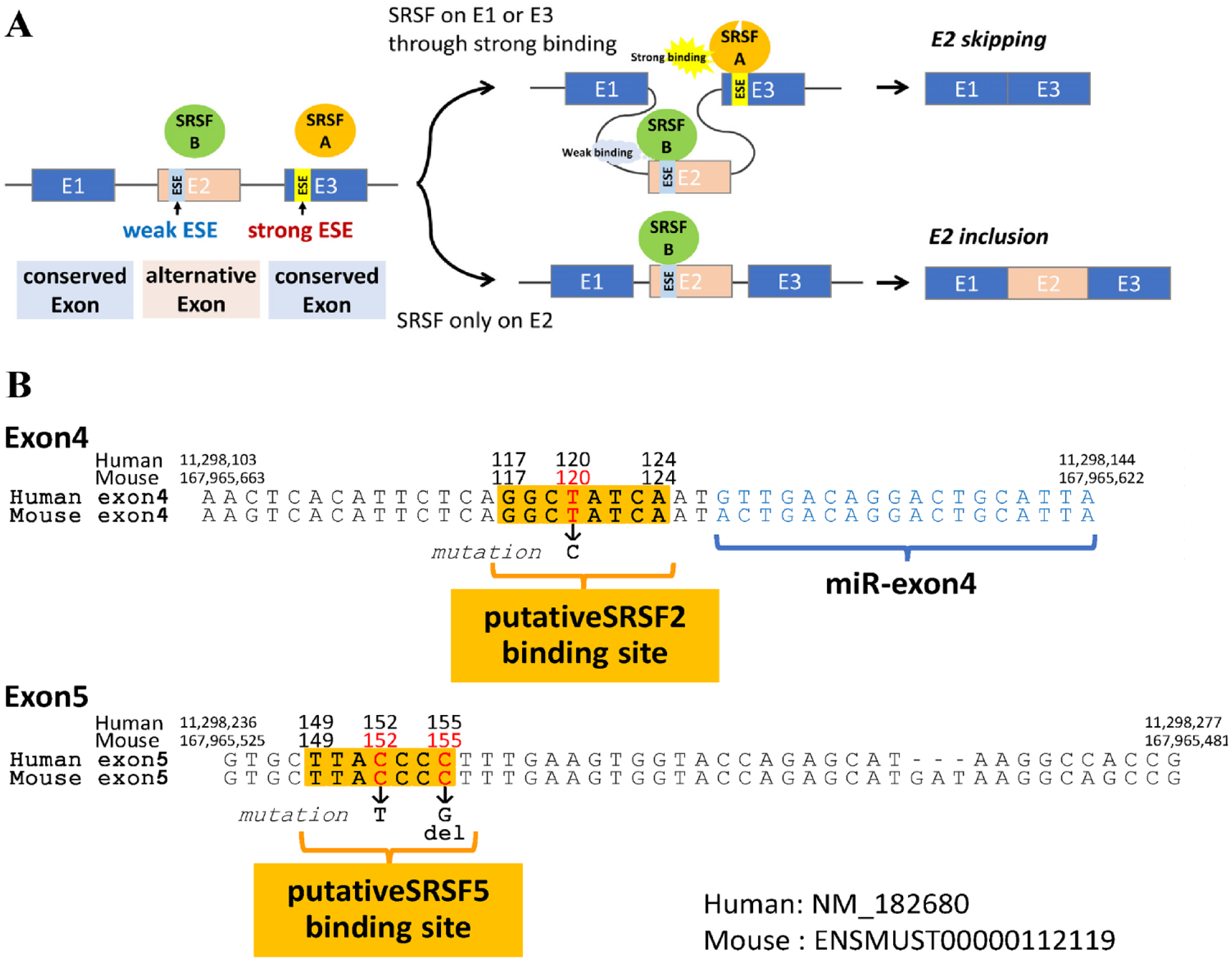
Alternative splicing and exon splicing enhancers (ESEs). (
In the case of amelogenin, exon4 is an alternative exon, and exon3 and exon5 are the conserved exons. In humans, point mutations (c.120T>C, and c.143T>C) located on a putative ESE sequence in exon4 result in X-linked amelogenesis imperfecta (AI) (Cho et al. 2014; Kim et al. 2020). This ESE is suggested for SRSF2 and SRSF6 binding, respectively, and the mutations are predicted to increase the SRSFs’ binding, resulting in more inclusion of exon4 in mRNA. Thus, these ESEs are supposed to be weak ESEs on the alternative exon. For exon4 to be alternatively spliced out, an interaction of the strong ESE and a respective SRSF is required on the conserved exons (i.e., exon3 or exon5).
In this study, we aimed to find the mechanisms regulating alternative splicing of exon4 to produce miR-exon4 and how the mutations causing X-linked AI affect this process using the amelogenin minigene, which is designed to understand the molecular mechanism of exon-intron splice site selection in the translated region of the amelogenin gene (Shapiro et al. 2006).
Materials and Methods
Animal
Mixed-background (C57BL/6J × SJL) mice were maintained at the UCSF animal facility in compliance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. The UCSF Animal Care Committee approved the animal handling and experimental protocol. Mice were euthanized at postnatal days 0 and 5 (P0 and P5, n = 4/each), and their first molars were harvested for total RNA extraction. At postnatal day 21, some mice were euthanized, and mandibles were dissected, fixed in 4% paraformaldehyde, decalcified, embedded in paraffin, and sectioned.
Amelogenin Minigene Mutagenesis
Amelogenin minigene vector plasmid (gifted by Dr. Michael Paine, University of Southern California) contains 5.75 kbp mouse amelogenin genomic DNA sequences from exon2 start codon to exon7 stop codon, generating alternative splice variants and miR-exon4 in LS8 cells (Shapiro et al. 2006; Le et al. 2016). QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) was used to introduce the mutations c.120T>C, c.152C>T, c.155C>G, and c.155delC.
Cell Culture and RNA/miRNA Extraction
HEK-293 cells and LS8 ameloblast-like cells (Chen et al. 1992) were plated at a cell density between 70,000 and 250,000 cells/cm2 and cultured with Dulbecco’s modified Eagle’s medium + GlutaMAX (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. After 24 h, minigene plasmid DNA was transfected into the cells with Lipofectamine 2000 (Thermo Fisher Scientific). After 24 h, cells were harvested, and RNA was extracted using the Direct-zol RNA miniprep plus kit (Zymo Research). miRNA was selectively transcribed into complementary DNA (cDNA) using miScript II RT kit (Qiagen), while mRNA was reverse transcribed using the Superscript IV First-Strand Synthesis System (Thermo Fisher Scientific).
Quantitative Polymerase Chain Reaction
Gene expression was analyzed by quantitative polymerase chain reaction (qPCR) using specific kits depending on the amplification’s necessity (Le et al. 2016). miR-exon4 was analyzed with the miScript SYBR Green PCR Kit (Qiagen) using Hs_SNORD95 as a reference gene. Amelogenin mRNAs containing exon4, Nfia, and Prkch were examined using TB Green Premix Ex Taq II (Takara Bio). Exon2-PA28956 and exon2-exon6d were amplified using the KOD SYBR qPCR mix (Toyobo), and Srsf2 and Srsf5 expressions were examined using the FastStart Universal SYBR Green Master Kit (Roche Diagnostics). Mrpl19 reference was used for all mRNA qPCR. Custom-synthesized primer sequences are listed in Appendix Table 1. The expression levels of target genes were analyzed by the ΔΔCt method and calculated as a fold change compared with the control. The significance of differences was determined by independent Student’s t test or the multiple t tests with Bonferroni correction following 1-way analysis of variance (ANOVA) using Prism software (GraphPad Software) with P < 0.05 considered significant.
Immunohistochemistry
Mouse mandible sections were deparaffinized, processed for heat-mediated antigen retrieval, and blocked with 10% swine and 5% goat sera. The primary antibodies, rabbit anti-human SRSF2 (Thermo Fisher Scientific, #PA5-12402) and rabbit anti-human SRSF5 (United States Biological, #S55554-58), were incubated overnight at room temperature. A biotinylated swine anti-rabbit IgG F(ab′)2 fraction (Dako Cytomation, #E0431) was used as the secondary antibody, followed by incubation with ALPase conjugated streptavidin (Vector Laboratories). The immunoreaction was visualized with the Vector Red Alkaline Phosphatase Substrate Kit (Vector Laboratories) added levamisole (1 mM). Images of immunoreactions were captured using cellSens software (EVIDENT) to evaluate the staining.
Reverse Transcriptase Polymerase Chain Reaction and Gel Electrophoresis
Exon2-exon6d amplification was done by reverse transcriptase polymerase chain reaction (RT-PCR) using a Phusion High-Fidelity PCR kit (Thermo Fisher Scientific), and amplicons were separated by gel electrophoresis using E-gel 2% agarose (Thermo Fisher Scientific) and the 5% Mini-PROTEAN TBE Gel (Bio-Rad). Bands were scanned and visualized with the Odyssey XF scanner (Licor) and Empiria Software (Licor).
Electrophoretic Mobility Shift Assay
Cy5.5-labeled RNA oligonucleotide probes for exon4 (200 ng) and exon5 (150 ng) (Dharmacon) were incubated with recombinant human SRSF2 (Creative Biomart) and SRSF5 (Proteintech) (5 fmol each) using the Odyssey Infrared EMSA Kit (Licor). Nonlabeled RNA oligonucleotide probes (5 pmol) for each exon were used as competitors. After incubation, samples were loaded to the 5% Mini-PROTEAN TBE Gel (Bio-Rad) for the electrophoresis at 200 V for 20 min. Gels were imaged using an Odyssey DLx scanner (Licor) and analyzed using Image Studio software (Licor).
Results
Identification of ESE in Exon5 Associated with X-Linked AI
To find out the possible SRSFs for exon4 skipping, we completed an in silico analysis and predicted ESEs and their binding SRSFs in the mouse and human amelogenin gene using ESE-Finder Ver 3.0 (Cartegni et al. 2003). Comparing the known mutations for X-linked AI in exon4 and exon5, our analysis identified a sequence at position 149–155 (Fig. 1B and Appendix Table 2) in exon5 as an ESE site for SRSF5 that included the sites for all 4 known mutations in exon5 (Aldred et al. 1992; Lench et al. 1994; Lench and Winter 1995; Kida et al. 2007; Prasad et al. 2016) (Fig. 1B). Given the mutations, the sequence was no longer predicted as an ESE. The mutations happen in the first or last quadruplicate C sequence. Therefore, these 2 Cs seemed critical for this sequence’s ESE property. Agreeing with previous reports (Cho et al. 2014; Kim et al. 2020), positions 117–124 (Fig. 1B) and 138–143 in exon4 are also predicted as ESEs for SRSF2 and SRSF6. However, position 138–143 fell in the miR-exon4 sequence. Thus, it was excluded from this study. All predicted ESE sequences were 100% homologous between humans and mice.
X-Linked AI-Causing Mutations Change Exon4 Splicing and Consequent miR-Exon4 Production
We introduced mutations in the amelogenin minigene to examine the effect of the mutations causing X-linked AI on exon4 splicing and miR-exon4 production. c.120T>C disrupts position 114–124 ESE and 3 types of mutations that replace or delete the first or last quadruplicate C sequence in position 149–155 ESE (c.152C>T, c.155C>G, and c.155delC). They were transfected into HEK-293 cells, an epithelial cell line expressing negligible endogenous amelogenin mRNA (Appendix Fig. 1). Next, exon4 inclusion and miR-exon4 production were examined by qPCR. The transfection efficiency of each minigene was assessed by measuring amelogenin mRNA transcribed exclusively from minigene or total HEK-293 cells using a primer set of exon2 and PA28956 (outside of amelogenin sequence but before polyA; Shapiro et al. 2006) or exon2-6d, respectively. In both cases, only the minigene with c.155delC mutation showed similarly lowered transfection compared to the wild-type (WT) (Fig. 2A–H). Therefore, qPCR data for miR-exon4 and amelogenin mRNAs containing exon4 were normalized with the minigene-specific total amelogenin mRNA.
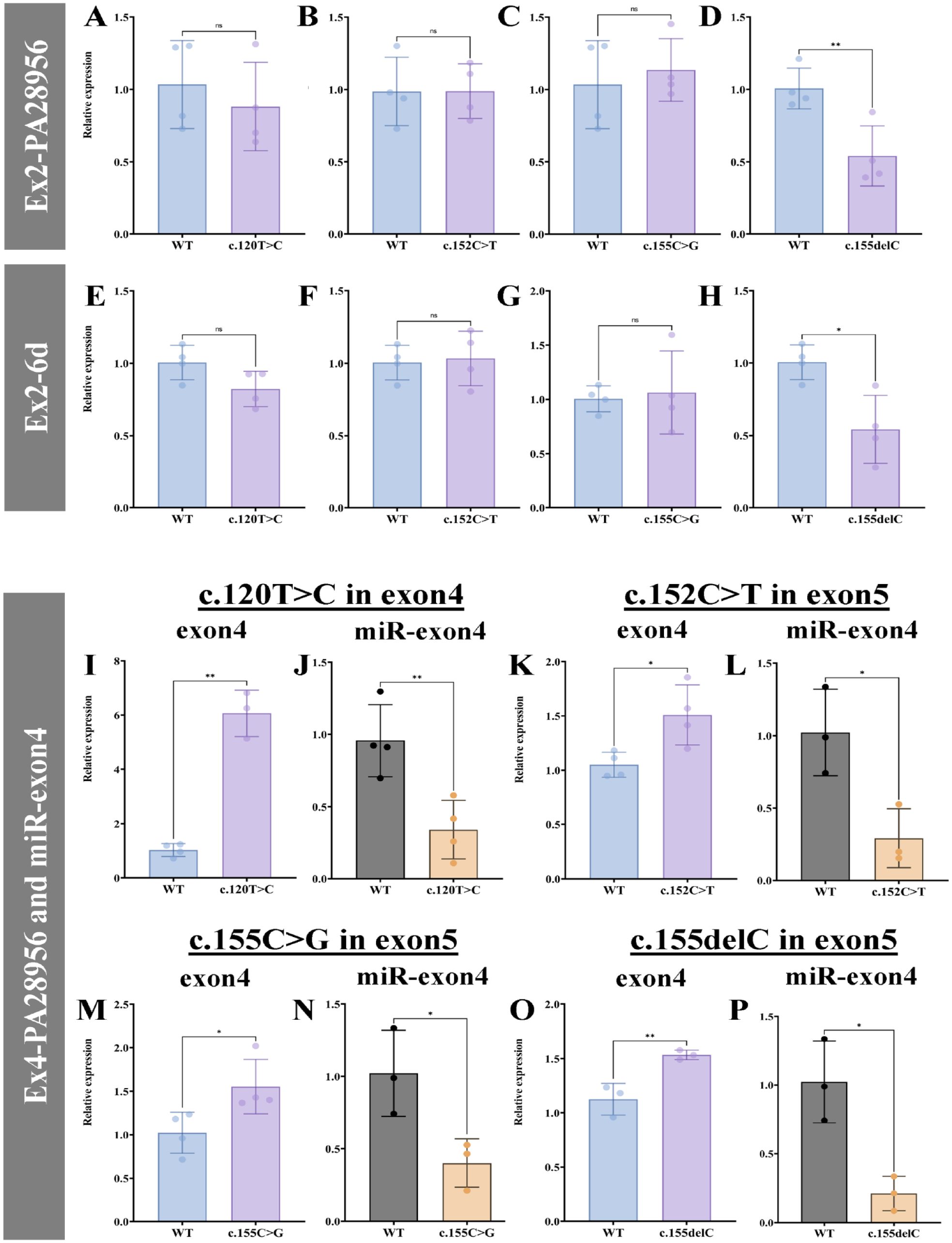
Quantitative polymerase chain reaction analysis of messenger RNA (mRNA) including exon4 and miR-exon4. (
Compared to the WT, all mutations significantly upregulated exon4 inclusion in the amelogenin mRNA (Fig. 2I, K, M, O). Simultaneously, miR-exon4 production was significantly downregulated with all mutations (Fig. 2J, L, N, P). This indicates that when more exon4 is included in amelogenin mRNA, less miR-exon4 is formed, suggesting the correlation between miR-exon4 formation and alternative splicing of exon4.
Amelogenin mRNA has 2 major splicing variants: long form, containing exons 2, 3, 5, 6abcd, and 7, and short form, containing exons 2, 3, 5, 6d, and 7. It gives distinct functions to the translated proteins, and in both cases, the inclusion of exon4 alters the function of the amelogenin proteins (Goldberg et al. 2009; Cho et al. 2014). To determine if the exon4 inclusion caused by the mutations has preferences between the 2 forms of amelogenin mRNA, we examined the mRNA variants derived from minigenes by gel electrophoresis. Exon4 inclusion in the bands was determined by pre–gel hybridization (Appendix Fig. 2). Changes in the intensity of bands containing exon4 (red arrows in Fig. 3A) were visually observed in all mutations for both long and short forms. As the RT-PCR products visualized on the gel did not reflect the long- and short-form ratio (Fig. 3B), we distinguished exon4 inclusion in each form by qPCR. All 4 mutations caused significantly higher exon4 inclusion in the long form compared to WT (Fig. 3C–F), while for the short form, 3 mutations (c.120T>C, c.155C>G, and c.155delC) caused significantly higher exon4 inclusion (Fig. 3G, I, J). The c.152C>T mutation rather caused slightly but significantly reduced exon4 inclusion in the short form (Fig. 3H).
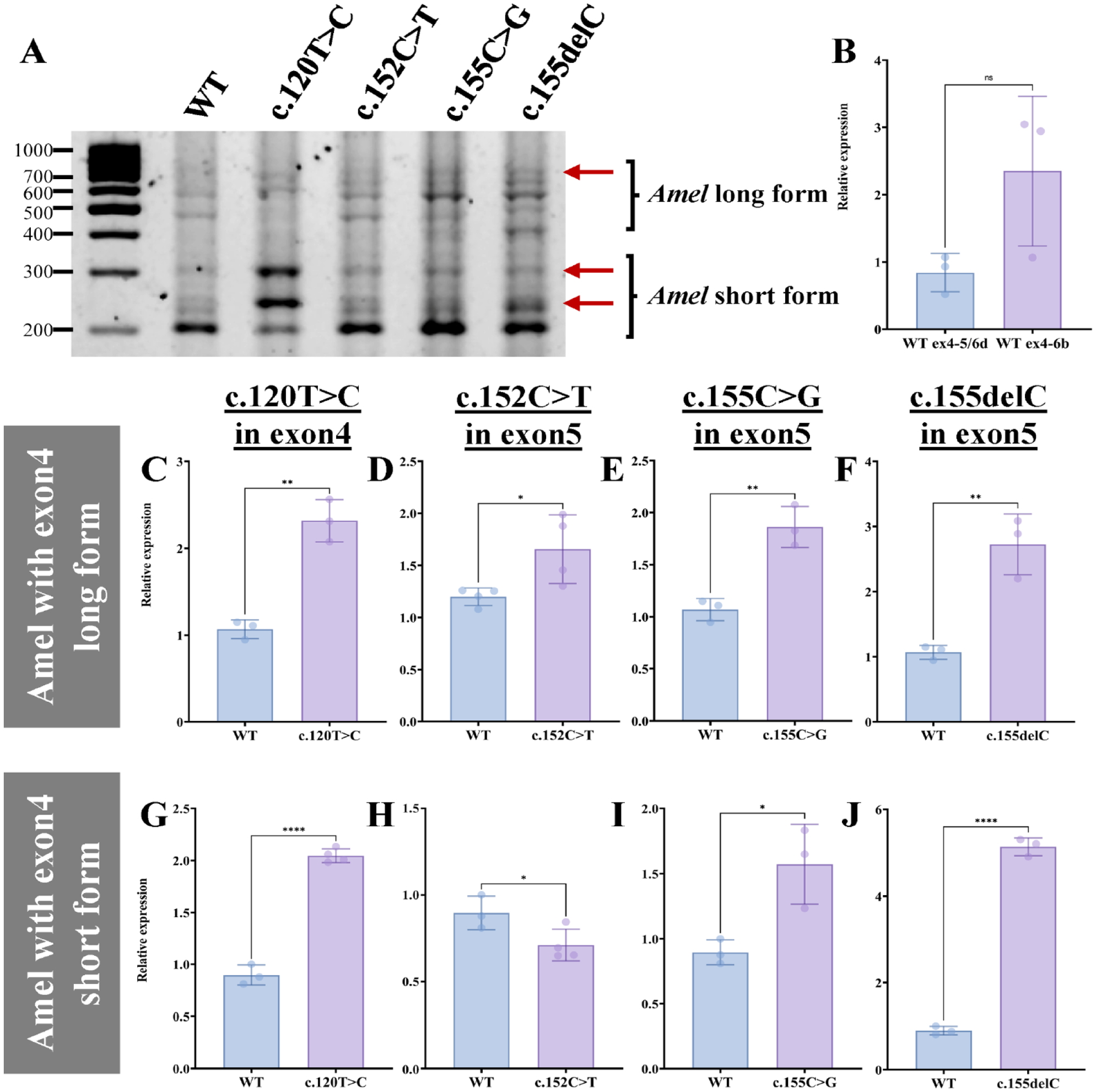
Exon4 inclusion in different forms of amelogenin messenger RNA (mRNA). (
X-Linked AI Causing Mutations on Exon4 and Exon5 Change SRSF Binding
To determine how the mutations altered exon4 splicing, we examined the binding of SRSF2 and SRSF5 to exon4 and exon5 and whether mutations alter SRSF binding to the putative ESEs by an electrophoretic mobility shift assay (EMSA). Incubating fluorescent-labeled exon4 WT RNA with SRSF2 formed 3 super-shifted bands of exon4-SRSF2 complexes (Fig. 4A). The super-shifted bands disappeared when the nonlabeled RNA competitor was added, confirming that SRSF2 specifically bounds exon4 WT. As the 3 complexes are different sizes, each complex was expected to have a different number of SFSR2 bound (detailed interpretation in Appendix Fig. 3). Increased super-shifted bands with c.120T>C mutation suggest more binding of SRSF2 to exon4 through the position 117–124 ESE.
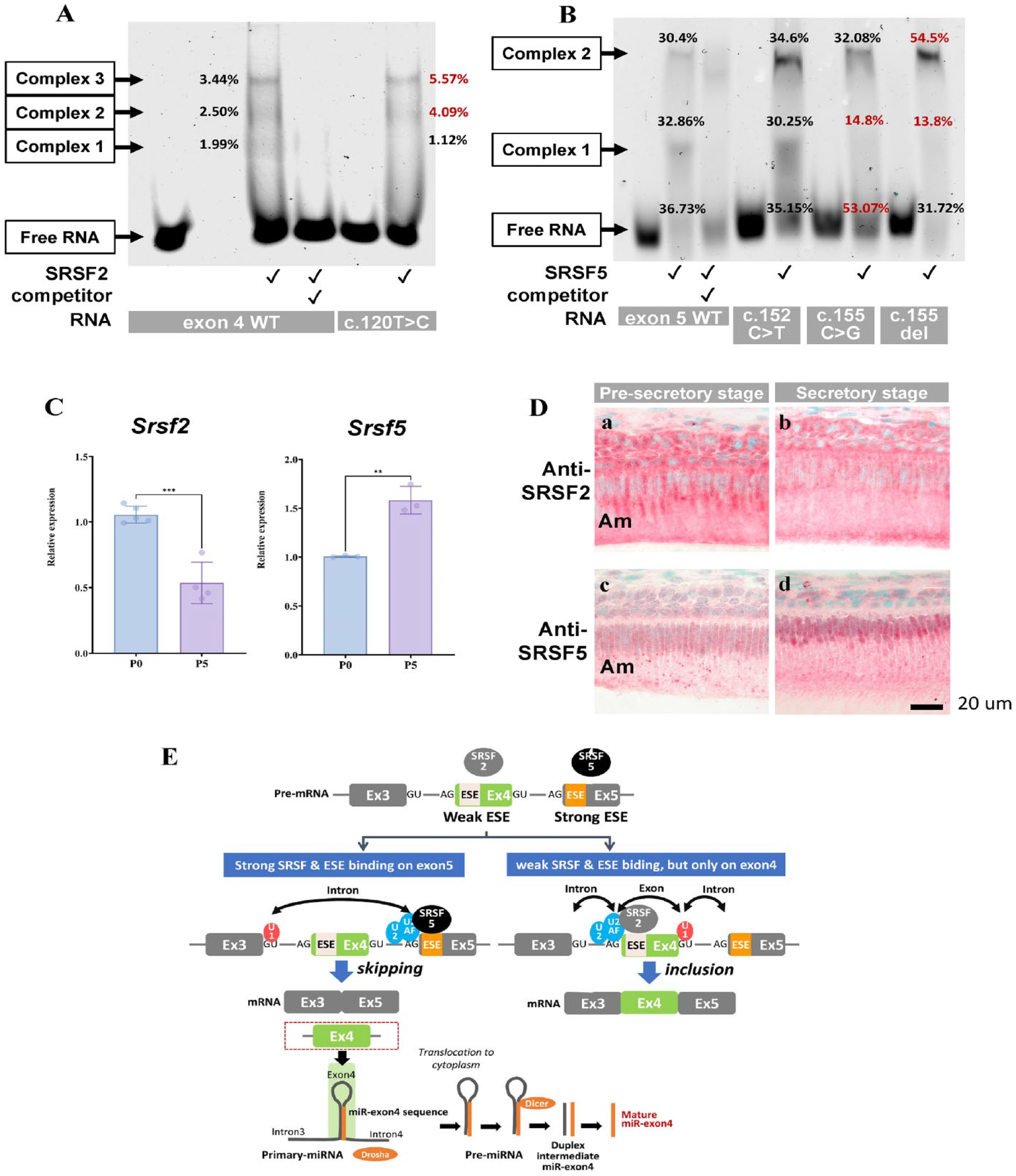
Ser/Arg-rich RNA splicing factors (SRSF) 2 and SRSF5 to regulate exon4 splicing. (
Similarly, incubation of exon5 WT RNA with SRSF5 showed the super-shifted bands (Fig. 4B). The disappearance of the super-shifted bands by adding a competitor confirmed SRSF5 and exon5 WT binding. As 2 complexes with different sizes, each complex was also expected to have a different number of SRSF5 bound to exon5 RNA (detailed interpretation in Appendix Fig. 4). Change in the super-shifted bands with the c.155C>G and c.155delC mutations indicated position 149–155 ESE is critical for SRSF5 and exon5 interactions, and the mutations disable the SRSF5 binding. The c.152C>T mutation did not change the ratio of SRSF5-exon5 complexes compared to WT.
To determine the SRSF2 and SRSF5’s role in amelogenin exon4 splicing in vivo, we detected Srsf2 and Srsf5 in mouse enamel organs. Amelogenin mRNA skips exon4 in about 80% of the case during ameloblast differentiation, and miR-exon4 expression greatly increases from presecretory to secretory stages (Le et al. 2016), suggesting the high capability of secretory ameloblasts to splicing exon4 than presecretory ameloblasts. Indeed, Srsf5 was significantly increased at P5/secretory stage compared to the P0/presecretory stage, while Srsf2 decreased (Fig. 4C). Immunostaining showed more nuclei in presecretory ameloblasts were positive for SRSF2 compared to the secretory ameloblasts (Fig. 4D-a and b), whereas more nuclei of secretory ameloblasts were positive for SRSF5 compared to the presecretory ameloblasts (Fig. 4D-c and d). All these data suggest that SRSF binding to exon4 and exon5 is the key for exon4 splicing to control the production of miR-exon4 (Fig. 4E). The 149–155 ESE on exon5 usually functions as a strong ESE for SRSF5 to navigate exon4 splicing and miR-exon4 production. The mutations on 149–155 ESE weaken SRSF5’s interaction with exon5, and mutation on 117–124 ESE on exon4 enhances SRSF2’s interaction with exon4. Both of these changes increase the interaction of SRSF2 and exon4, leading to more inclusion of exon4 and reduced miR-exon4 production.
The Potential Effect of Mutations on miR-Exon4 Targets
An miRNA can suppress target mRNA’s translation and cause decay of the target mRNA (Llave et al. 2002; Linsley et al. 2007; Arvey et al. 2010; Hausser and Zavolan 2014). We previously reported that the miR-exon4 mimic downregulated expression of its direct targets, Prkch and Nfia, in LS8 cells (Shemirani et al. 2022). To examine if the minigene-derived miR-exon4 also alters these direct targets, we measured the expression of Prkch and Nfia in the LS8-transfected minigene with the mutations. Transfecting amelogenin WT minigene moderately but significantly reduced Prkch expression (Fig. 5A). c.155C>G and c.155delC mutations significantly upregulated the Prkch expression compared to the WT (Fig. 5B). Nfia expression was significantly suppressed by transfection of the WT minigene (Fig. 5C), but the mutations did not further alter the expression compared to the WT (Fig. 5D).
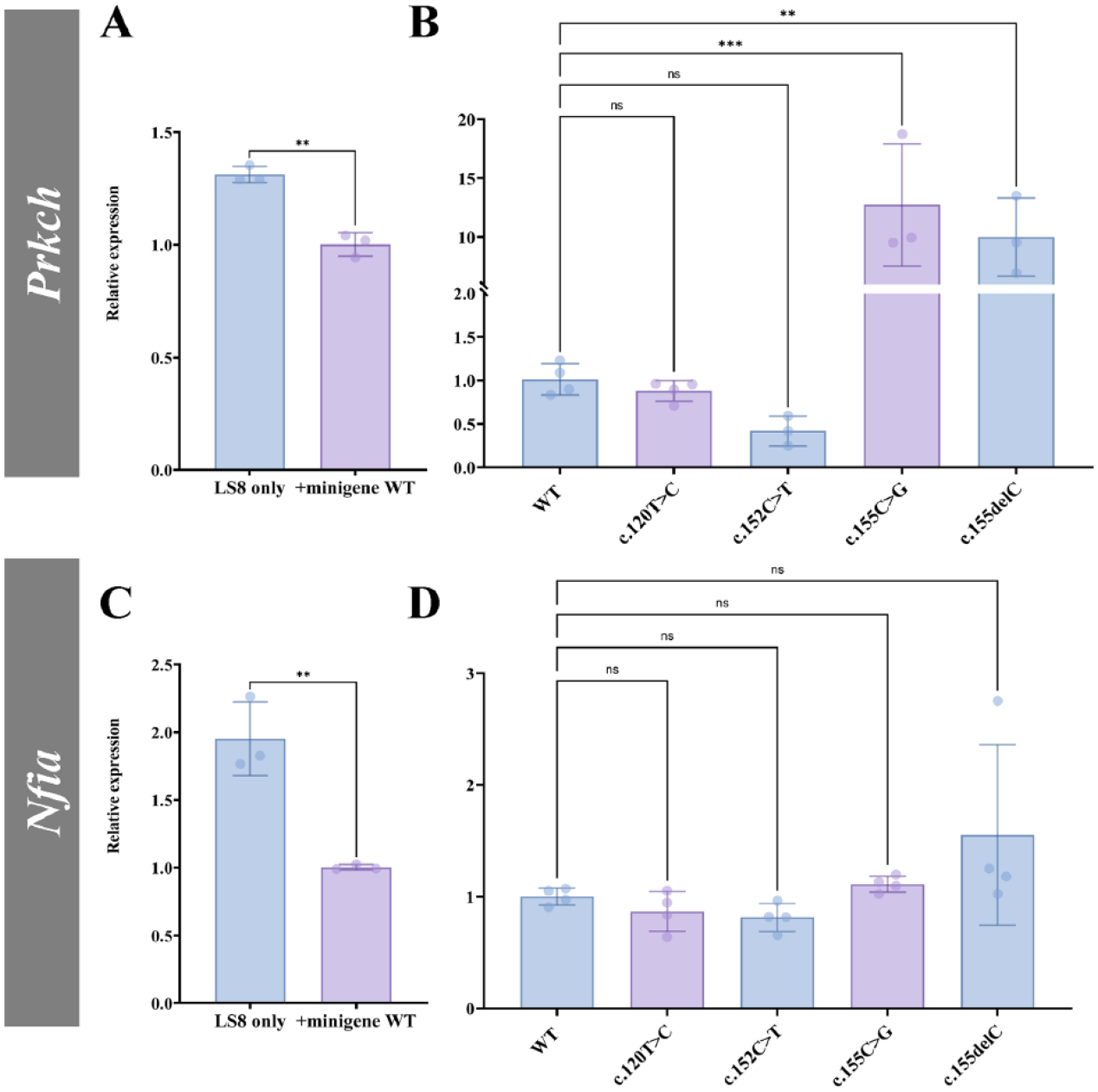
Expression of miR-exon4 direct targets in LS8 cells. (
Discussion
In this study, we identified the putative strong ESE in exon5 that SRSF5 could target in association with the mutations causing X-linked AI. We found that the mutations in the ESEs in exon4 and exon5 significantly changed amelogenin exon4 splicing and miR-exon4 production. Furthermore, we demonstrated that the SRSF2 and SRSF5 binding to the ESEs in exon4 and exon5 is the one way to regulate the alternative splicing of exon4 and consequent miR-exon4 production.
Alternative splicing of mRNA is a powerful means to produce multiple proteins from a single gene, occurring in over 90% of genes to provide functional complexity in higher organisms (Johnson et al. 2003; Wang et al. 2008). Greater than 60% of human disease-causing mutations affect the splicing patterns of genes rather than directly affecting coding sequences (Lim et al. 2011). Although it is well known that mutations in the amelogenin gene cause X-linked AI, alternative splicing has not been considered associated with the etiology of AI until recently (Cho et al. 2014; Grodecka et al. 2014; Kim et al. 2020).
Among the pathologic mutations examined in this report, all exon5 mutations are expected to cause altered protein coding, p.Thr51Ile, p.Pro52Arg, and p.Pro52Leufs*2, while exon4 mutation is a silent mutation (Aldred et al. 1992; Lench et al. 1994; Lench and Winter 1995; Kida et al. 2007; Cho et al. 2014). Besides the traditional protein coding, this report shows that mutations causing human X-linked AI can change the alternative splicing of exon4, leading to increased amelogenin protein containing exon4 and reduced miR-exon4 production. In mice, exon4 amelogenin mRNAs are detected in ameloblasts throughout its differentiation (Le et al. 2016), while as a protein, amelogenin with exon4 is detected mostly in the maturation stage (Stahl et al. 2015), suggesting its significant role of exon4 coding amelogenin, particularly in enamel maturation. Overexpressing amelogenin long form with exon4 throughout the enamel formation in mice, including the secretory stage, resulted in enamel defects similar to the human X-linked AI (Cho et al. 2014), indicating the strict stage-specific control of exon4 inclusion under physiologic conditions.
Also, distinct effects of the short form of amelogenin (LRAP) with or without exon4 were shown on dentin formation and differentiation of odontoblasts and ameloblasts (Tompkins et al. 2005; Lacerda-Pinheiro et al. 2006; Ye et al. 2006). Thus, the definite meaning of exon4 is suggested for LRAP-driven signaling in enamel formation. Our data showing that the mutations increased exon4 inclusion for both long and short forms of amelogenin indicate that the mutations in exon4 and exon5 affect both functions of amelogenin, an enamel matrix protein and a signaling molecule. According to the literature, patients with those mutations do not show specific phenotypes other than those commonly seen in X-linked AI patients. This would occur because the altered 2 major amelogenin proteins can cause significant phenotypes regardless of whether alteration occurs due to the amino acid sequence or insertion of exon4. Interestingly, the c.152C>T mutation did not change the exon4 inclusion in the short form of amelogenin while increasing in the long form, suggesting an association between exon4 and exon6abc splicing. With the c.152C>T mutation, exon4 inclusion is possibly enhanced, but less exon6abc is spliced. Therefore, 149–155 ESE in exon5 acts as a site for multiple splicing regulators, and the C at position 152 would be critical to coordinate exon4 and exon6abc splicing. In support of this possibility, the c.152C>T mutation did not change the SRSF5 binding, further suggesting the presence of some other SRSF(s) that can regulate both exon4 and exon6abc splicing.
As a new type of amelogenin derivative from alternative splicing, miR-exon4 is also suggested to have a significant signaling role in ameloblasts and osteoblasts (Shemirani et al. 2022), although the amelogenin minigene is not ideal for observing the downstream signaling effect due to the cytomegalovirus promoter. Here in this study, we observed downregulation of miR-exon4’s direct target (Prkch and Nfia) by the amelogenin minigene, which provides excessive miR-exon4 (Le et al. 2016), and further upregulation of Prkch associated with the c.155C>G and c.155delC mutations. The pattern of the direct target expression follows the principles of miRNA and targets. However, the reaction did not happen with all mutations and all targets. It is obviously due to the influence of the simultaneous presence of minigene-derived excessive amelogenin proteins as the signaling regulator in the LS8 ameloblastic cells. Also, each mutation creates different effects on the amelogenin protein coding and possibly another alternative splicing pattern. Therefore, it is highly likely that the miR-exon4 and amelogenin proteins synergistically work to regulate the differentiation and activities of ameloblasts. The altered miR-exon4 expression is suggested as an additional possible etiology for enamel defects seen in X-linked AI.
Author Contributions
R. Shemirani, Y. Nakano, contributed to conception and design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; M.H. Le, contributed to conception and design, data acquisition, analysis, and interpretation, critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345231180572 – Supplemental material for Mutations Causing X-Linked Amelogenesis Imperfecta Alter miRNA Formation from Amelogenin Exon4
Supplemental material, sj-docx-1-jdr-10.1177_00220345231180572 for Mutations Causing X-Linked Amelogenesis Imperfecta Alter miRNA Formation from Amelogenin Exon4 by R. Shemirani, M.H. Le and Y. Nakano in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Institute of Dental and Craniofacial Research, National Institutes of Health (grant no. 5R01DE027366). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.