
Abstract
Periodontitis is a common complex inflammatory disease of the oral cavity. It is characterized by inflammation of gingival tissues and alveolar bone loss. Recently, a genome-wide association study and 2 genome-wide association study meta-analyses found 2 associated regions (haplotype blocks) at the inhibitory immune receptor gene SIGLEC5 to increase the risk for periodontitis. The aims of the current study were the identification of the putative causal variants underlying these associations, characterization of their molecular biological effects, and validation of SIGLEC5 as the target gene. We mapped the associated single-nucleotide polymorphisms to DNA elements with predictive features of regulatory functions and screened the associated alleles for transcription factor (TF) binding sites. Antibody electrophoretic mobility shift assays (EMSAs) with allele-specific probes were used to identify TF binding and to quantify allele-specific effects on binding affinities. Luciferase reporter assays were used to quantify the effect directions and allele-specific strength of the associated regulatory elements. We used CRISPR-dCas9 gene activation to validate SIGLEC5 as a target of the association. EMSA in peripheral blood mononuclear cells showed that E-26 transformation–specific TF-related gene (ERG) binds at rs11084095, with almost complete loss of binding at the minor A-allele. Allele-specific reporter genes showed enhancer function of the DNA sequence at rs11084095, which was abrogated in the background of the A-allele. EMSA in B lymphocytes showed that TF MAF bZIP (MAFB) binds at the common G-allele of rs4284742, whereas the minor A-allele reduced TF binding by 69%, corresponding to 9-fold reduction of luciferase reporter gene activity by the A-allele. Using CRISPR-dCas9, we showed that the enhancer at rs4284742 strongly activated SIGLEC5 expression, validating this gene as the target gene of the association. We conclude that rs11084095 and rs4284742 are putatively causal for the genome-wide significant associations with periodontitis at SIGLEC5 that impair ERG and MAFB binding, respectively.
Keywords
Introduction
Periodontitis is a common complex inflammatory disease of the oral cavity and is characterized by chronic inflammation of the gingiva in conjunction with destruction of connective tissue and, subsequently, alveolar bone and tooth loss. Periodontitis has a range of manifestations that differ among individuals in severity and progression of tissue destruction and age of disease onset. The basis of phenotypic variation of complex diseases largely is the genetic variability among individuals (Timpson et al. 2018). The genetic variants mostly are single-nucleotide polymorphisms (SNPs; 1000 Genomes Project et al. 2015), and the number of functional SNPs and their biological consequences, magnitude of effects, and interactions with one another and with environmental factors shape disease manifestations (Boyle et al. 2017; Wray et al. 2018). To identify genetic susceptibility loci of periodontitis, various genome-wide association studies (GWASs) have been performed. Recently, a GWAS and 2 GWAS meta-analyses independently found SNPs at the inhibitory immune receptor gene sialic acid–binding Ig-like lectin 5 (SIGLEC5) to be associated at a genome-wide significance level (P < 5 × 10-8) with early-onset generalized stage III, grade C periodontitis (Munz et al. 2017) and more moderate late-onset forms (Munz et al. 2019; Shungin et al. 2019). Additionally, a study that integrated expression quantitative trait locus (eQTL) data with GWAS associations implied SIGLEC5 as periodontitis risk gene (Li et al. 2020). All SNPs of these associations are located in the introns of SIGLEC5. These are regions that do not code for a protein, but variants in introns can change the activity of a gene or cause a protein to be produced in the wrong place or at the wrong time. The closest genes in a distance of the associated SNPs are SIGLEC14 and ZNF175. eQTL effects for rs4284742 and rs11084095 suggested SIGLEC5 as the target gene of the association, with the most significant eQTL effects detected in blood and monocytes, the tissue where SIGLEC5 is almost exclusively expressed (except of placenta), with P = 7.7 × 10–14 and P = 6.4 × 10–23, respectively. Reported eQTLs of the associated SNPs were much weaker for other genes, implying SIGLEC5 as the most likely target gene of the association. SIGLEC5 is an inhibitory transmembrane receptor that binds sialic acids and sialic acid–containing glycan ligands, which are expressed on the surfaces of certain pathogens and bacteria. It is discussed that it mediates crosstalk of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) during infection and wound healing (Pillai et al. 2012).
To elucidate the molecular mechanisms that predispose to increased disease susceptibility, genetic associations need to be leveraged to biological meaning. This poses a challenge because the most significant associated variant, called the GWAS lead SNP or sentinel variant, most often is not identical with the functional variants that caused the association. This is explained by the fact that numerous SNPs are in strong linkage disequilibrium (LD) and coinherited with the GWAS lead SNPs, comprising associated haplotype blocks (Gabriel et al. 2002).
The aims of the current study were to identify the putative causal variants of the associations with SIGLEC5, to characterize the role of the effect alleles in the disease etiology, and to validate SIGLEC5 as the target gene of the association.
Methods and Materials
Protocols for cell culture, transfection, quantitative reverse transcription polymerase chain reaction, isolation of peripheral blood mononuclear cells (PBMCs), and eQTL analysis are given in the appendix.
Selection of Putative Causal Variants
To identify putative causal variants, we integrated data from ENCODE (ENCODE Project Consortium 2012) with the genomic locations of the associated SNP (Appendix). We determined all SNPs in strong LD (r2 > 0.8) to the GWAS lead SNPs (Munz et al. 2017; Shungin et al. 2019) in the northwestern European populations CEU (Utah residents with northern and western European ancestry) and GBR (British in England and Scotland; 1000 Genomes Project et al. 2010) of the International Genome Sample Resource using the online tool LDproxy (Machiela and Chanock 2015). Subsequently, sequence to motif alignments for these SNPs were performed with various libraries of binding matrixes for transcription factor binding sites (TFBSs) from Transfac professional (geneXplain), SNPInspector (Genomatix), and the open access database Jaspar. The transcription factor (TF) binding motif was confirmed via the web interface for position weight matrix (PWM) model generation and evaluation PWMTools (Ambrosini et al. 2018).
Electrophoretic Mobility Shift Assay
To determine allele-specific protein-DNA binding, the Gelshift Chemiluminescent EMSA Kit (Activemotif) was used. The nuclear protein extract of Raji cells was extracted as described in the Appendix. For the supershift binding reaction, Raji cells or PBMC nuclear extract (10 µg) and biotin-labeled double-stranded oligonucleotides (20 fmol) were incubated at room temperature for 20 min with 1× binding buffer and 2 µL of specific antibody. To verify the result of DNA-protein interaction, unlabeled oligonucleotides (4 pmol) were added to the binding reaction. The reactions were loaded onto a 5% native polyacrylamide gel and run in 0.5× TBE buffer at 100 V for 1 to 1.5 h. After electric transfer of the products to a nylon membrane, the membrane was cross-linked at 120 mJ/cm2; the biotin-labeled oligonucleotides were detected by chemiluminescence; and the absolute value area of the shifted bands was quantified by ImageJ. All electrophoretic mobility shift assays (EMSAs) were performed in triplicates. Sequences of oligonucleotide probes, antibodies, and analysis details are given in the Appendix.
Luciferase Reporter Gene Assay
Cloning and transfection of the luciferase reporter gene experiments are described in the appendix. All transfections were performed in 3 independent biological replicates.
CRISPR-dCas9 Activation
CRISPR-dCas9 activation (CRISPRa) provides the possibility to test whether a genomic site serves as a cis-regulatory element for a target gene of interest (Simeonov et al. 2017). It allows specific and efficient quantification of the regulatory potential that a chromatin sequence has on gene expression in the endogenous context, including naturally occurring variants. We used CRISPRa to analyze if the DNA elements at rs4284742 and rs11084095 had regulatory effects on SIGLEC5 expression. Details are given in the Appendix.
Results
Assignment of Putative Causal Associated Variants by Integration of LD and Regulatory Chromatin Features
The haplotype block tagged by the GWAS lead SNPs were relatively narrow and encompassed few intronic cosegregating SNPs (Fig. 1A, B). The closest genes in a distance of the associated SNPs are SIGLEC14 and ZNF175 (Appendix Fig. 1). rs4284742 (Munz et al. 2017) had no other variant in strong LD (r2 > 0.8), whereas the adjacent associated haplotype block that was tagged by the GWAS meta-analyses’ lead SNPs rs12461706 (Shungin et al. 2019) and rs11084095 (Munz et al. 2019) comprised 5 SNPs in strong LD. These included the 2 sentinel variants and 3 additional tagging SNPs (Table). Of the 6 SNPs that tagged the 2 haplotype blocks, only rs4284742 mapped to regulatory DNA elements as determined by ChIP-Seq and DNAse I hypersensitivity. rs11084095, rs34984145, and rs11880807 located to a region that showed H3K4Me1 methylation in the B-cell line GM12878, a methylation mark that is enriched at active and primed cell type–specific enhancers. Both haplotype blocks were separated from one another by an insulator element (Fig. 1C).
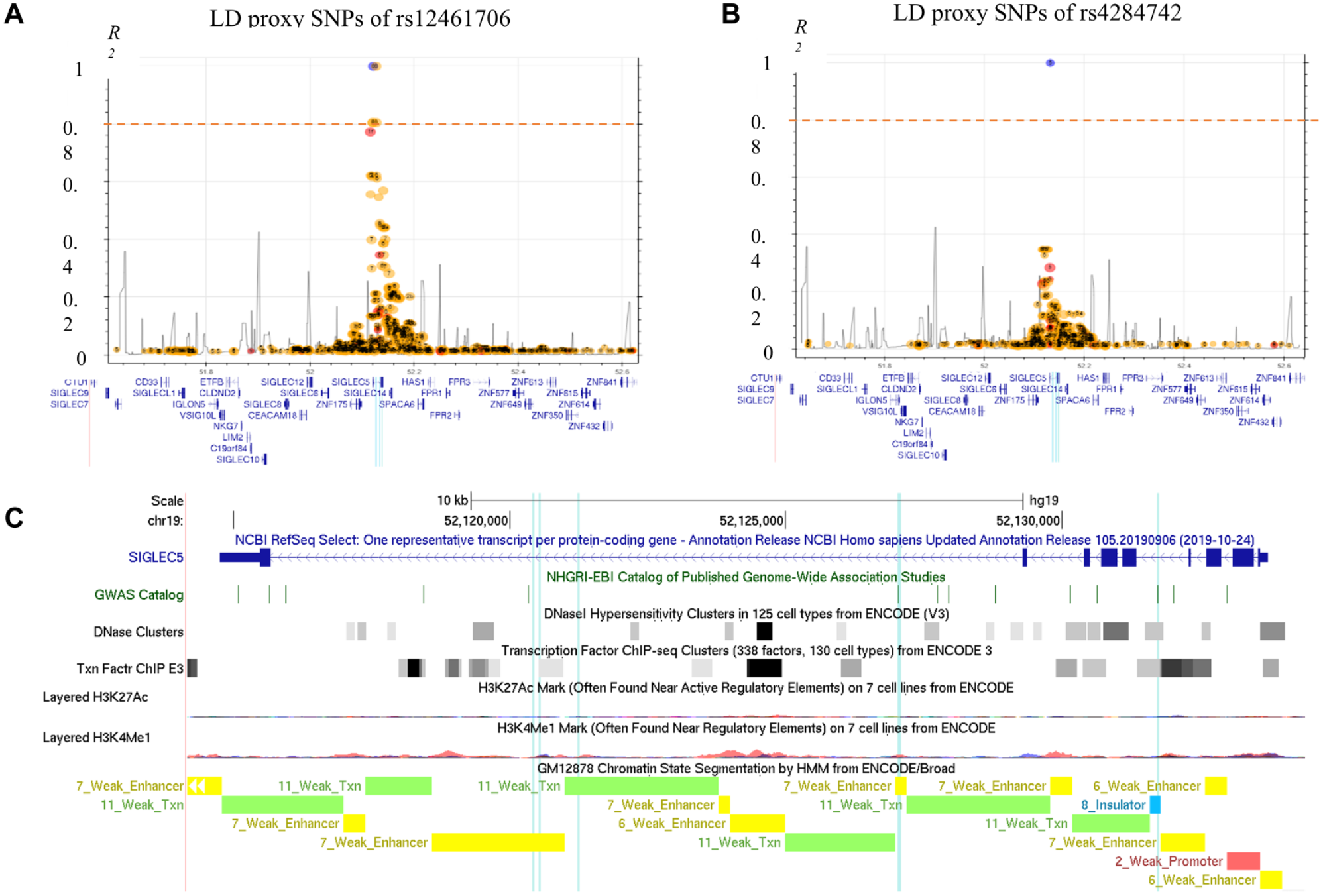
Linkage and chromosomal positions of the single-nucleotide polymorphisms (SNPs) that showed genome-wide significant associations with increased periodontitis susceptibility. (
SNPs in Linkage Disequilibrium (r2 > 0.8) with the GWAS Lead SNPs rs4284742, rs12461706, and rs11084095.
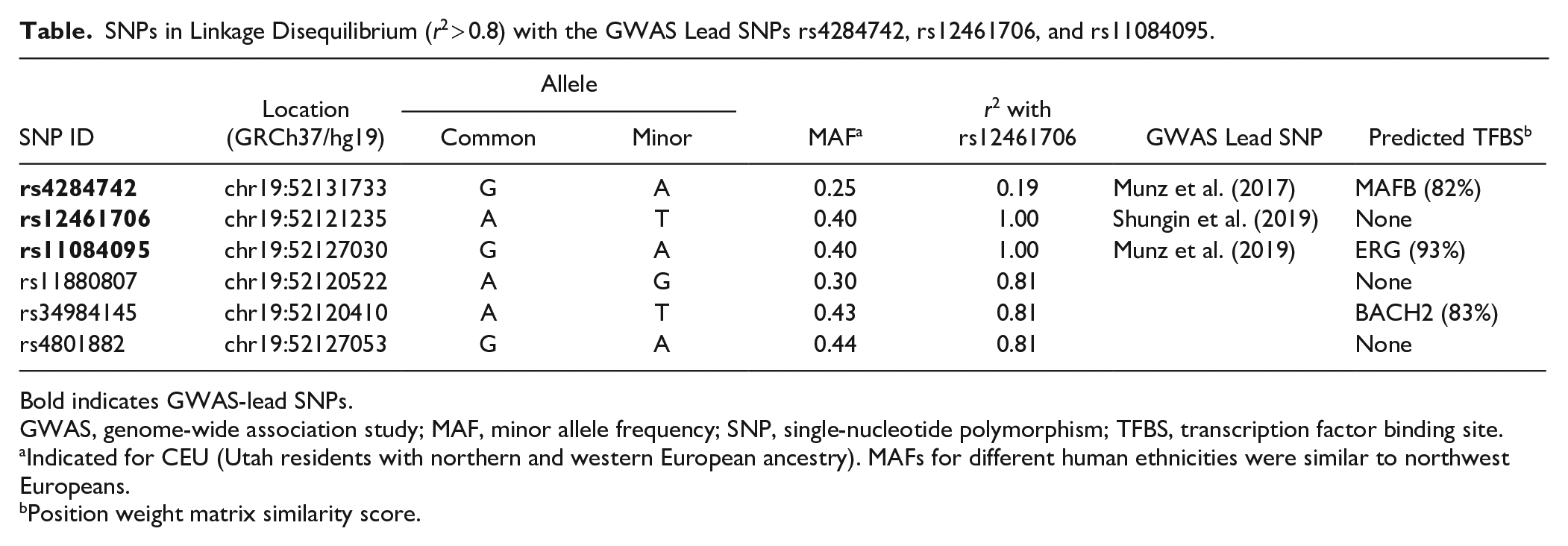
Bold indicates GWAS-lead SNPs.
GWAS, genome-wide association study; MAF, minor allele frequency; SNP, single-nucleotide polymorphism; TFBS, transcription factor binding site.
Indicated for CEU (Utah residents with northern and western European ancestry). MAFs for different human ethnicities were similar to northwest Europeans.
Position weight matrix similarity score.
We investigated PWM libraries whether the nucleotide variants of the 6 SNPs changed predicted TFBSs. For the common allele of rs11084095, a TFBS for the ETS transcription factor ERG with a matrix similarity of 93% was predicted (Fig. 2A); for the common allele of rs4284742, a TFBS for the TF MAF bZIP transcription factor B (MAFB) with a matrix similarity of 82% was predicted (Fig. 2B); and for the common allele of rs34984145, a TFBS for BACH2 with a matrix similarity of 83% was predicted (Fig. 2C). PWMTools confirmed the high similarity to the TF binding motifs at these SNPs. At the noneffect alleles of the other SNPs, no TFBS was predicted. We selected the 3 SNPs rs4284742, rs11084095, and rs34984145 for subsequent validation of TF binding in vitro.
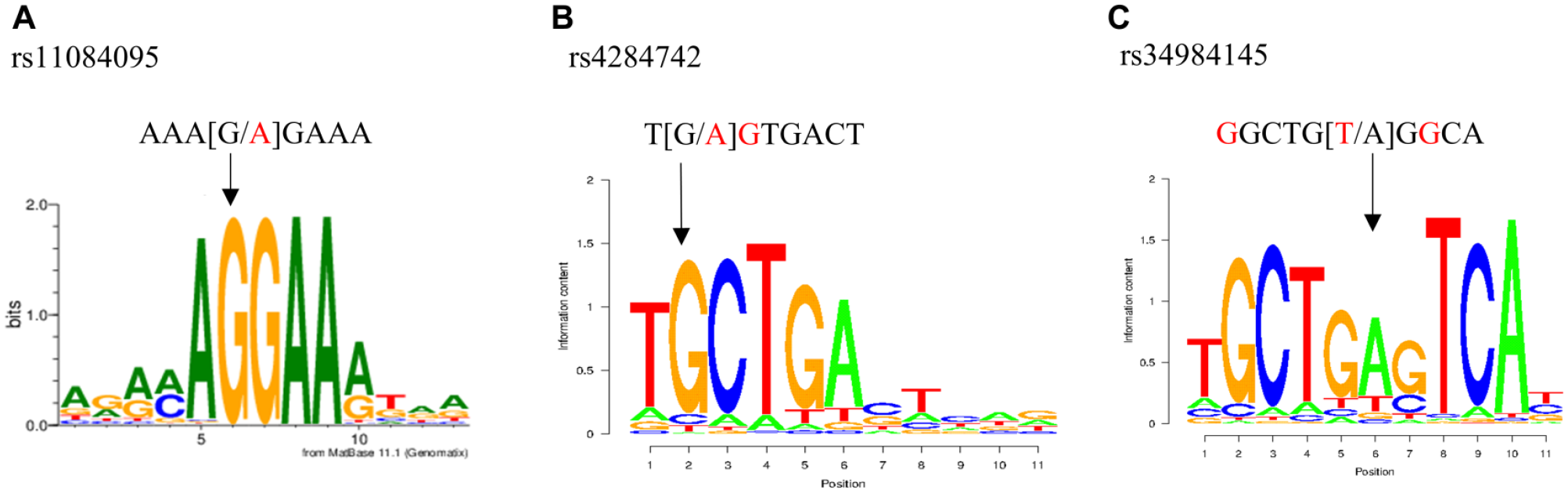
rs11084095 and rs4284742 have allele-specific effects on ERG and MAFB binding. (
TFs ERG and MAFB Show Allele-Specific Binding at rs11084095 and rs4284742
To prove protein binding at the 3 selected SNPs and to give evidence for allele-specific binding of the predicted TFs, we performed an EMSA using ERG, MAFB, and BACH2 antibodies with allele-specific DNA probes. SIGLEC5 is mainly expressed in various lymphocytes of the innate immune system and in B cells (Crocker and Varki 2001). We performed the EMSA for rs11084095 with protein extract from PBMCs because in addition to SIGLEC5, ERG is weakly expressed in this cell type, too. MAFB is expressed in whole blood (including B lymphocytes), and BACH2 is expressed in B lymphocytes. Therefore, we performed the EMSA for rs4284742 and rs34984145 with protein extract from a B-lymphocyte cell line (Raji cells). In each EMSA we observed a band supershift with the TF-specific antibody and allele-specific oligonucleotides (Fig. 3A–F). The background of the rare A-allele of rs11084095 significantly reduced ERG binding with P = 0.005 to 99% as compared with binding at the common G-allele (Fig. 3A, B). The background of the rare A-allele of rs4284742 significantly reduced MAFB binding with P = 0.02 to 69% compared to binding at the common G-allele (Fig. 3C, D). At the common A-allele of rs34984145, BACH2 binding showed a significant reduction with P = 0.007 of TF binding by 59% as compared with the rare T-allele (Fig. 3E, F).
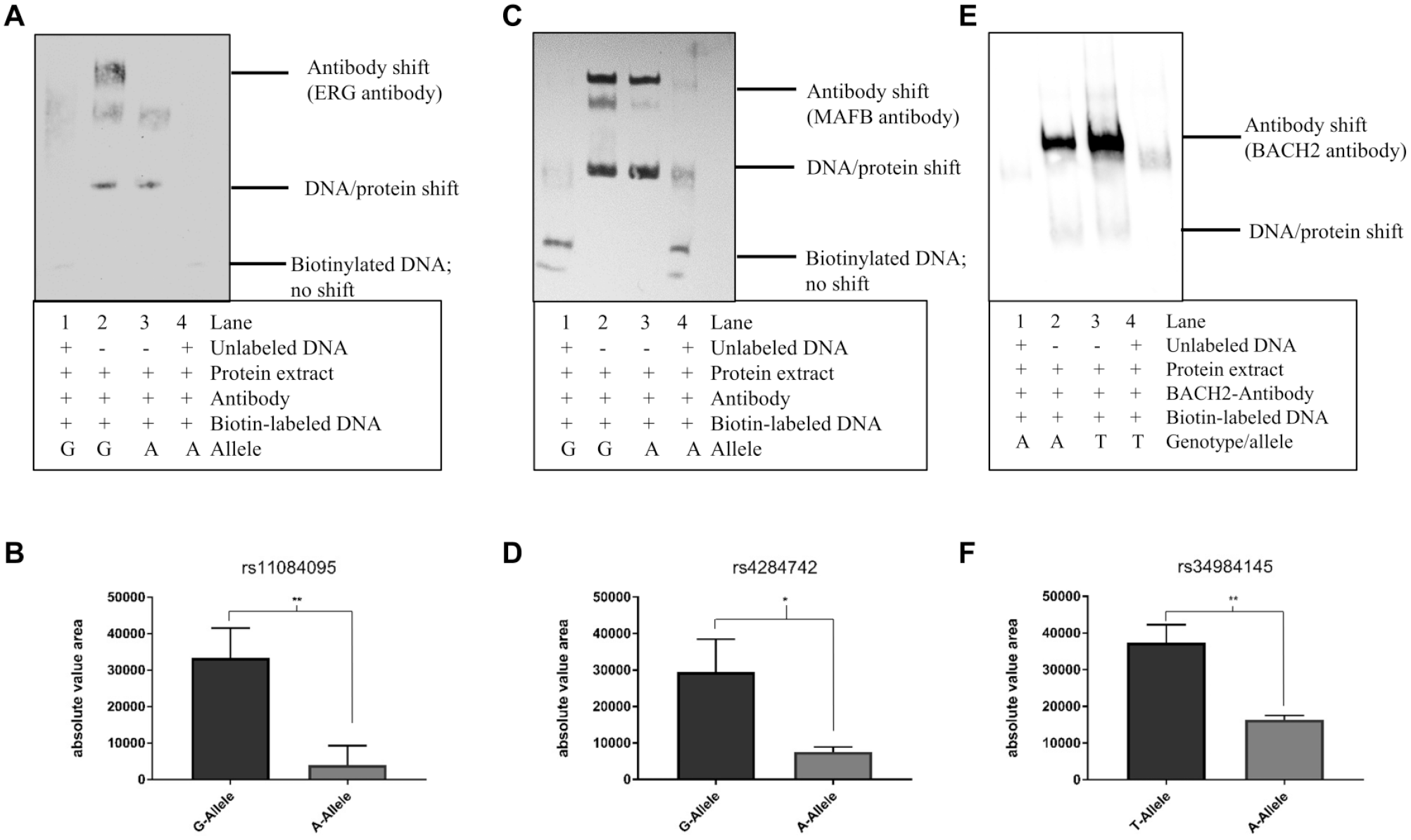
Transcription factor binding site of BACH2 at rs34984145. (
rs4284742, rs11084095, and rs34984145 Are Located in Transcriptional Activators
Regulatory DNA elements can either activate or repress transcription. To measure the activity of the regulatory elements at rs11084095, rs4284742, and rs34984145 and to discriminate their effect directions and allele-specific effect sizes, we employed luciferase reporter gene assays. ERG is expressed in HeLa cells at similar levels as in PBMCs. Because HeLa cells can be transfected more efficiently than PBMCs, which improves the detection of differences in reporter gene expression, we performed the reporter gene experiments in HeLa cells. Here, luciferase activity showed a significant increase in the background of the common G-allele of rs11084095 by 9.9-fold (P = 0.015) as compared with the empty plasmid. The rare A-allele showed no increase in luciferase activity. The difference between alleles was significant with P = 0.013 (Fig. 4A).
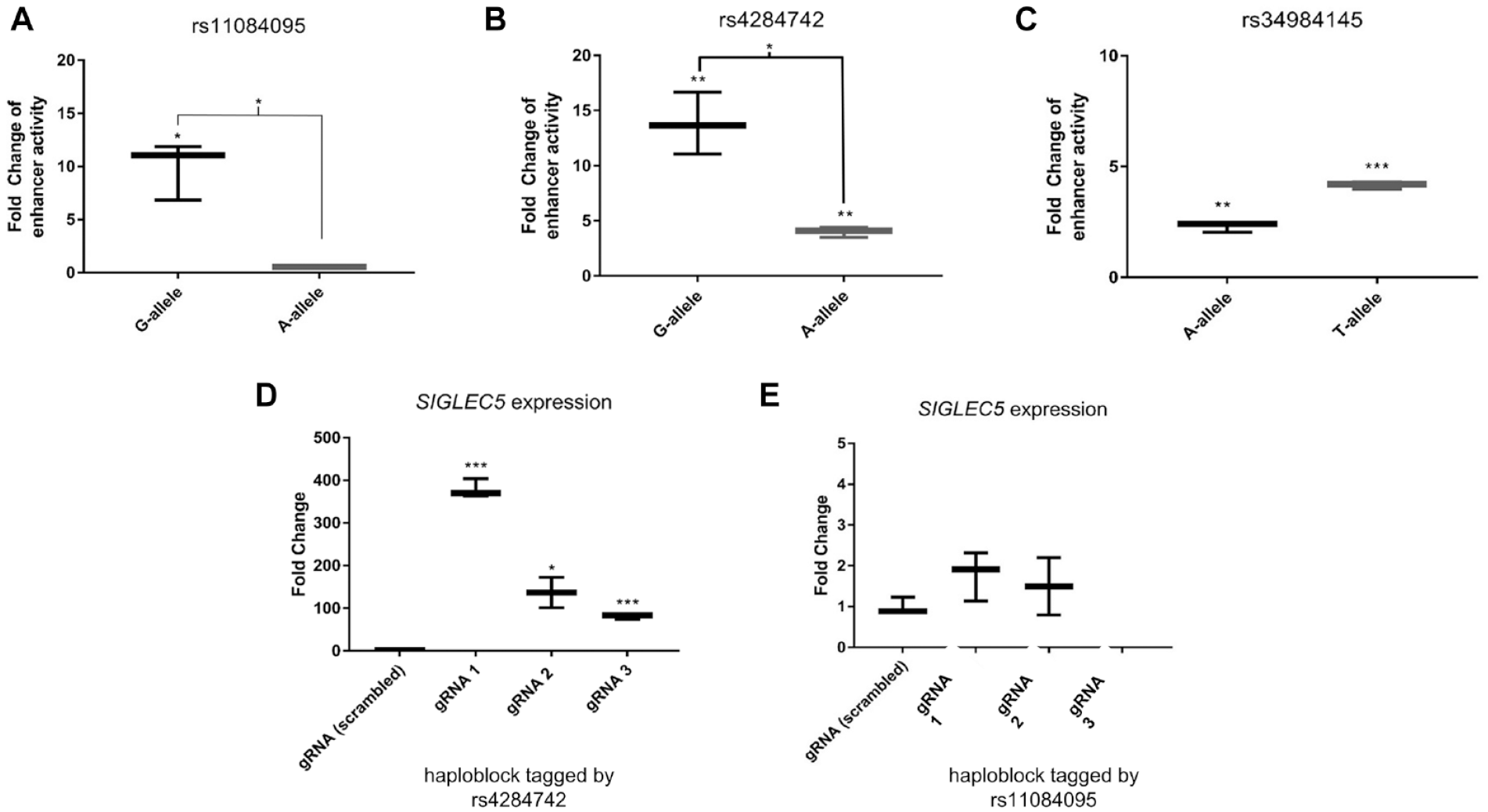
rs11084095 and rs4284742 have allele-specific effects on luciferase activity. The 65-bp DNA sequence up- and downstream of rs11084095 and the 75-bp DNA sequence spanning rs4284742 showed allele-specific enhancer activity in HeLa cells. (
The DNA sequence with the common G-allele of rs4284742 showed 13-fold upregulation of luciferase activity (P = 0.008) in HeLa cells as compared with the reporter gene without this regulatory element (Fig. 4B). The reporter gene with the minor A-allele showed an upregulation of 4-fold (P = 0.002). The difference between alleles was significant with P = 0.01.
The luciferase gene activity of rs34984145 showed weak but significant upregulation for both alleles (T-allele: 4.2-fold, P = 0.0001; A-allele: 2.3-fold, P = 0.002), but the fold change difference between alleles was <2 (Fig. 4C), indicating allele-specific effects of low biological relevance.
These experiments indicated that the regulatory elements act as enhancers. Additionally, these experiments showed strong allele differences of rs11084095 and rs4284742 on enhancer activity.
CRISPR-dCas9 Activation of rs4284742 Showed Cis-regulation of SIGLEC5
For rs11084095, an eQTL effect on the expression of SIGLEC5 was reported in monocytes with P = 6.4 × 10–23 (Zeller et al. 2010), and for rs4284742, an eQTL effect on the expression of SIGLEC5 was noted in whole blood with P = 7.7 × 10–14 (Blood eQTL browser; Westra et al. 2013), suggesting that the disease-associated elements have cis-regulatory effects on the expression of SIGLEC5 (Appendix Tables 1 and 2). To validate the regulatory potential of the associated haplotype blocks tagged by rs11084095 and rs4284742, we performed CRISPRa in HeLa cells because Raji cells showed poor survival after transfection with the CRISPRa plasmids, probably because of DNA toxicity (Kim et al. 2014). CRISPRa of rs4284742 (gRNAs located in 20- to 300-bp distance from the SNP) with the synergistic activation mediator (SAM) system strongly increased the expression of SIGLEC5 as compared with cotransfected unspecific control sgRNAs (FC = 380, Fig. 4D; Appendix Fig. 2). This proved that the DNA element at rs4284742 exerts a strong cis-regulatory effect on SIGLEC5 and implied that SIGLEC5 is the target gene of the disease-associated genetic variant. CRISPRa of rs11084095 with 3 gRNAs (25- to 120-bp distance from the SNP) showed no significant upregulation of SIGLEC5 (Fig. 4E).
Because the alleles of rs34984145 showed no different effects on luciferase activity and no bloodborne eQTLs of rs34984145 on the expression of SIGLEC5 were reported, we considered that rs34984145 is not a causal variant of the association and did not perform CRISPRa for this SNP.
Discussion
Two large GWASs independently found genetic variants at SIGLEC5 to be associated with different forms of periodontitis, indicating broad relevance of this locus for the disease etiology. We identified rs4284742 and rs11084095 as putative causal variants of the associations with early-onset generalized stage III, grade C periodontitis (Munz et al. 2017) and with more moderate forms (Shungin et al. 2019), respectively. Both SNPs were not in LD (r2 = 0.2) and located on different haplotype blocks. However, rs12461706 was in complete LD (r2 = 1) with rs11084095, the lead SNP of a GWAS-meta-analysis that combined the generalized stage III, grade C periodontitis cases with a GWAS sample of more moderate periodontitis (Teumer et al. 2013), which was also included in the GWAS meta-analysis of Shungin et al. (2019). To identify the putative causal variants of these associations, we analyzed all SNPs that were in strong LD to the GWAS lead SNPs for locating to predicted TFBSs. The 2 complementary TF databases of Transfac professional and SNPInsepctor allowed prediction of binding sites of all human TFs currently known. However, it is possible that we missed a TF with no currently known binding site. Another possible limitation was that we confined the analysis to SNPs that indicated strong linkage according to r2 > 0.8. We used this LD measure because the r2 coefficient of correlation takes account of allele frequency. Strong LD indicated by D′ but not by r2 would include alleles that are inherited with the particular GWAS lead SNP but are not carried by the majority of cases because they are rare. Such alleles would not be suggestive as causative variants because they would not explain the association for most cases. However, this does not exclude the existence of rare susceptibly variants at SIGLEC5, but such variants have no disease-relevant role for the general population. Instead, their effects would become noticeable in individual cases.
At the position of rs11084095, we identified a binding site for the TF ERG with 93% PWM matrix similarity. We showed that the sequence at rs11084095 is a binding site for ERG and has enhancer activity. The background of the rare effect allele significantly reduced ERG binding and enhancer activity. ERG is an essential TF for endothelial homeostasis, a system control state that encompasses acute responses to injury to support repair of damaged endothelium (Heiss et al. 2015). Changes in endothelial homeostasis (i.e., endothelial injury) affect the vascular structure by interacting with extracellular matrix turnover and influence endothelial membrane function and adhesiveness to proteins of the coagulation cascade and platelets, membrane permeability, and integrity (Gulino-Debrac 2013). Corresponding to function, ERG is mainly expressed in endothelial cells but also in leukocytes (Uhlen et al. 2015). The GWAS catalog (Welter et al. 2014) reports that genetic variations of ERG are associated with numerous blood cell traits and blood pressure, as well as bone mineral density and osteoarthritis. Notably, it was recently shown that soluble SIGLEC5 appears efficient in blocking leukocyte rolling over P-selectin (platelet selectin) and E-selectin (endothelial selectin; Pepin et al. 2016). In human endothelial cells, ERG associates with enhancers of von Willebrand factor (Kalna et al. 2019) and von Willebrand factor may act as a ligand for SIGLEC5 (Pegon et al. 2012), suggesting a possible link between endothelial homeostasis and SIGLEC5 function.
At the position of SNP rs4284742, we identified a predicted TFBS of the TF MAFB and showed that the effect allele, which is the common allele, provided strong MAFB binding affinity as compared with the noneffect allele. Additionally, we showed that CRISPR-dCas9 activation of the genomic sequences at rs4284742 activated SIGLEC5 expression. These experiments indicated that MAFB regulation is linked with activation of SIGLEC5 expression and with increased risk for early-onset periodontitis. MAFB belongs to the subfamily of the large Maf transcription factors (Santos-Gallego 2016). It is expressed by monocytes and is required for differentiation to macrophages (Kelly et al. 2000; Gemelli et al. 2006). Furthermore, it negatively regulates osteoclast generation via inhibition of the transcription factor NFATc1 and the osteoclast-associated receptor (OSCAR; Kim et al. 2007), implying a functional context of the genetic association with osteoclast differentiation. Additionally, MAFB was shown to promote sprouting angiogenesis (Jeong et al. 2017). During healing of tissue injuries, angiogenic capillary sprouts invade the wound clot and organize into a microvascular network throughout the granulation tissue (reviewed by Tonnesen et al. 2000).A challenge for healing of aseptic tissue injuries is discrimination from infections. This is achieved by the innate immune system through danger- or pathogens-associated molecular patterns, DAMPs and PAMPs. Sialoside-based pattern recognition by SIGLEC5 receptors was shown to selectively suppress the immune response to DAMPs, suggesting a mechanism by which aseptic tissue injury and infection are distinguished (Chen et al. 2009). This mechanism is normally well controlled to avoid autoimmune destruction. However, many viruses and pathogenic bacteria express sialidase as virulence factors (Crennell et al. 1993; reviewed by Drzeniek 1972; Crennell et al. 1993), and microbial-expressed sialidases might have potential to abrogate the SIGLEC-mediated inhibitory effects of DAMPs on the innate immune system during healing of injured tissues. As a result, DAMPs and PAMPs would become indistinguishable, which could provide an explanation for massive inflammation and tissue destruction (Liu et al. 2009), as typically seen in severe periodontitis phenotypes (stage III) with a rapid rate of progression (grade C). This may explain why, in specific situations, SIGLEC5 expression may increase the risk for alveolar bone loss.
Notably, the maximal PWM score of MAFB was 82%. We interpret the incomplete similarity score in a way that the specificity of protein-DNA binding depends not solely on the DNA sequence but also on the 3-dimensional structure of DNA and TF protein macromolecules (Rohs et al. 2010). This result in variation of functional binding motifs and, accordingly, the predicative accuracy of a PWM should be interpreted with caution (Weirauch et al. 2013). However, the rare allele further reduced the matrix similarity to 60%, which corresponded with reduced MAFB binding in the EMSA and reduced reporter gene activity. Using CRISPRa, we could show that the enhancer at rs4284742 regulates SIGLEC5 expression. However, we lacked experimental evidence for such effects at rs11084095. This does not signify that SIGLEC5 is not the target gene of this association. Notably, the strongest eQTL of rs11084095 affected SIGLEC5 expression (e.g., P = 6.4 × 10–23 in monocytes; Zeller et al. 2010) but no other genes. We consider technical problems that impeded the function of the CRISPRa system at this haplotype block—for example, poor targeting or binding of the gRNAs at the selected PAM sequences.
For the common allele of rs34984145, a TFBS for BACH2 was predicted with a PWM matrix similarity of 83%. The antibody-specific EMSA gave evidence for BACH2 binding at the sequence of this SNP, but the luciferase reporter gene showed no differences of enhancer activity between alleles. This is why we considered that rs34984145 is not a causal variant of the association.
We conclude that rs4284742 and rs11084095 are functional variants, that the risk alleles reduce enhancer activity and binding affinity of the TFs MAFB and ERG, and that SIGLEC5 is the target gene of these associations. We suggest a functional context of the associations with impaired regulation of endothelial homeostasis and healing of aseptic tissue injuries.
Author Contributions
R. Mueller, A.S. Schaefer, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; A. Chopra, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; H. Dommisch, contributed to data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345211049984 – Supplemental material for Periodontitis Risk Variants at SIGLEC5 Impair ERG and MAFB Binding
Supplemental material, sj-docx-1-jdr-10.1177_00220345211049984 for Periodontitis Risk Variants at SIGLEC5 Impair ERG and MAFB Binding by R. Mueller, A. Chopra, H. Dommisch and A.S. Schaefer in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the German Research Foundation (SCHA1582 5-1).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.