
Abstract
Novel fillers from sago starch and esterified sago starch were employed as natural fillers for rigid polyurethane foams (RPUFs). Sago starch was esterified by maleic anhydride, resulting in the presence of ester groups on the starch structure. Filled RPUFs were prepared with 0.5-7.0 wt% of starch fillers in the polyol component. The influence of filler type and content on the cell morphology and properties of the RPUFs was analyzed. The results revealed that the esterified sago starch showed better compatibility with polyurethane matrix than the sago starch, which in turn impacted the cellular morphology and physico-mechanical properties of the resulting RPUFs. The density and compressive strength of the RPUFs filled with esterified sago starch were higher than those filled with unmodified sago starch, while their water absorption, and volume shrinkage were lower. The findings also suggested that the compressive strength and density of filled RPUFs increased with starch filler content up to the optimal point and then decreased. This was due to the impact of filler content on cell size, with smaller cell size at low filler content leading to increased strength and density, whereas larger cell size and more open cells at higher filler content reducing strength and density. The best properties were obtained with 1.0 wt% of unmodified sago starch and 0.5 wt% of esterified sago starch in the polyol component.
Introduction
Rigid polyurethane foams (RPUFs) are low-density porous polymers with high insulation properties, making them suitable for a range of applications such as insulations for refrigerators, panels, piping, tanks, and roofing.1,2 Enhancing the properties of RPUFs has been the focus of extensive research, with a particular emphasis on incorporating optimal amounts and types of fillers into the foam structure. There have been several reports claimed that the employ of various types of fillers e.g. organofluorine, 3 bio and bio-derivative fillers such as: walnut shells and silanized walnut shells, 4 wood flours, 5 microcrystalline cellulose, 6 potato protein, 7 nanofillers, for example: cellulose nanocrystals, 8 montmorillonite, 9 carbon nanotubes 10 act as nucleation sites and thus reduce the cell size and, as a result, effectively increase the mechanical properties, fire resistance and thermal stability and reduce the thermal conductivity of RPUFs.9,10
With increasing concern for sustainability and the pursuit of sustainable development goals (SDGs), incorporating natural fillers into polyurethane foams presents a potential approach to improve mechanical and insulating properties,8,11 decrease non-renewable petrochemical demand, and limit carbon dioxide emissions. 12 Hence, the trend towards modifying RPUFs with fillers derived from renewable, bio-based sources has grown in recent years.
Starch, a versatile biomaterial, is of significant interest for various food 13 and non-food applications. 14 Starch non-food applications include biomembranes as drug delivery system phamaceuticals, 15 textiles, 16 alcohol-based fuels, 17 adhesives, 18 dyes, and packaging19–21 due to its abundance, low cost, non-toxicity, and biodegradability. 22 Sago starch, derived from the sago palm (Metroxylon sagu Rottb.), is a particularly dominant socioeconomic crop in Southeast Asia that remains underutilized. The structure and properties of sago starch have been previously documented.23–25
Previous reports have shown that sago starch has a high amylose content (15%–35% by weight,24,25 resulting in high crystallinity and superior mechanical properties compared to other starches. As a result, sago starch has been explored as a filler for linear low density polyethylene. 19 Our previous work also successfully prepared a thermoplastic film from sago starch. 20
Despite its potential for use in polymeric materials, sago starch has not been previously considered as a filler for RPUFs due to its limited compatibility with the reactive liquid starting materials of isocyanates and polyols. To overcome this limitation, the starch crystallinity was decreased by modifying sago starch with maleic anhydride. 26 This modification was expected to improve compatibility with the liquid starting materials and enhance reactivity of hydroxyl group on sago starch and isocyanate, leading to improved functional properties and promoting environmental sustainability.
Experimental
Materials
The polymeric 4,4-diphenyl methane diisocyanate (p-MDI) used in this study was sourced from Nippon Polyurethane Industry (Millionate MR200), Japan. Voranol RN490, a petroleum-based polyether polyol with a hydroxyl number of 493 mg KOH/g, was produced by Dow Chemicals Co. Ltd, Germany. The silicone surfactant (Niax Silicone L626) was obtained from Momentive Performance Materials, USA, the amine catalyst (Tegoamin 33) from Evonik Nutrition & Care GmbH, Germany, and dibutyl tin dilaurate, DBTDL (Aldrich, USA) were used as received. The sago starch filler was extracted from the sago palm plant, which contained 24% amylose (determined by an amperometric titration adapted from McCleary et al.). 27
Chemical modification of sago starch fillers
The modified sago starch was prepared through esterification with maleic anhydride as described in previous literature. 26 100 g of dry sago starch was blended with 5 g of maleic anhydride powder and homogenized in a conical flask at a controlled temperature of 80°C. The esterified starch was washed with acetone to eliminate any residual maleic anhydride and by-products, and dried in an oven at 50°C for 24 h.
Foam preparation
Rigid polyurethane foam formulations.
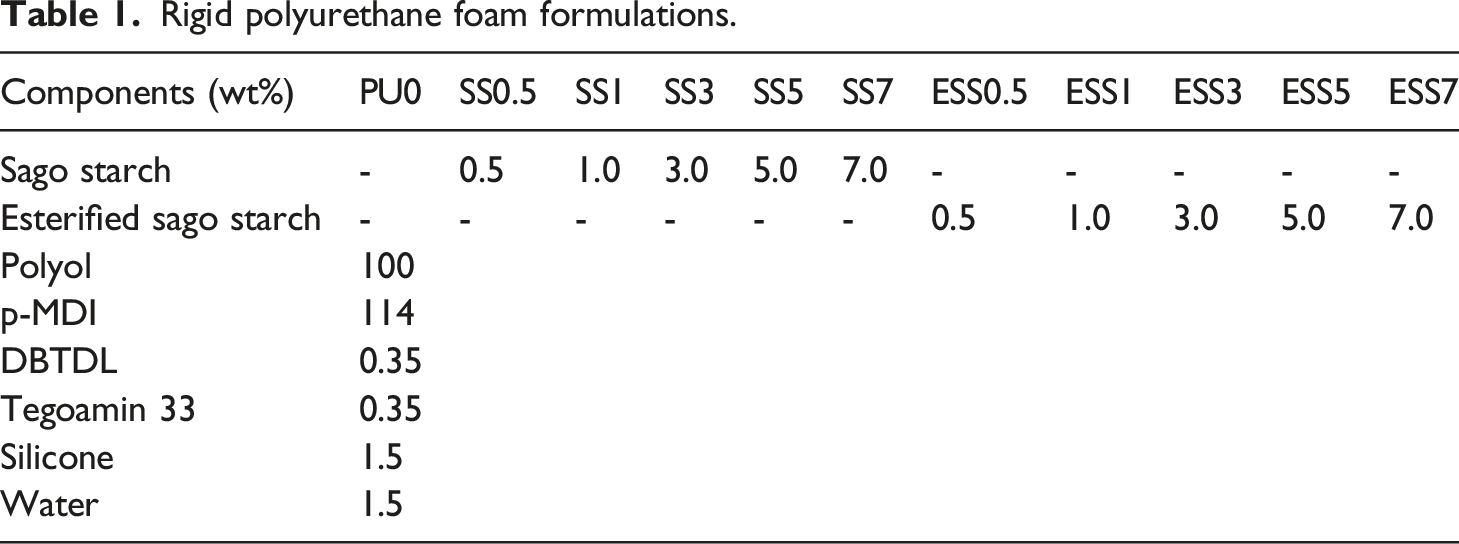
To make the RPUFs, the ingredients for the polyol mixture, which included polyol, catalysts, blowing agents, surfactant, and sago starch (Component A), were weighed out and placed in a plastic container. The mixture was mixed using an overhead stirrer at 1000 rpm for approximately 60 s. Component B, p-MDI, was then added to the polyol mixture and stirred for 30 s at 1000 rpm. The mixture was then allowed to expand freely in the container. The exothermic temperature changes during the foaming process were continuously monitored and recorded using a type J thermocouple with a data logger (DP-74SD, Digicon). The characteristic foaming times such as cream time (initiation of foaming), full rise time (full expansion of foaming), and tack-free time were recorded. The produced RPUFs were conditioned at room temperature for 24 h, and then cut into appropriate shapes using a band saw based on testing standards, after which their physical properties were investigated.
Characterization
The FTIR spectra were taken using a Tensor 27 instrument from Bruker, Switzerland and had a wavenumber range of 4000-400 cm−1 at a resolution of 4 cm−1.
The X-ray diffraction of starch was conducted using an X-ray diffractometer (model Panalytical – Empyrean, Netherlands), equipped with a Cu target (λ = 0.1540 nm), at room temperature. The diffractometer comprised a rotating anode generator operating at 35 kV and 20 mA, and the samples were scanned from 2θ = 5 to 40° in a step scan mode.
The viscosities of 7.0 wt% polyol-sago starch blends were measured using rheometer (HR20, TA Instruments, USA) with a cone-plate geometry. The upper plate consisted of a cone with a diameter of 40 mm and a cone angle of 2°. The viscosity was monitored as a function of shear rate at ambient temperature.
The shrinkage of 30 × 30 × 25 mm specimens was evaluated and calculated using the following equation:
The morphologies of starches and the cellular morphologies of the polyurethane composite foams were characterized using a LEO1455 scanning electron microscope (SEM), Germany. Rectangular samples, approximately 2.5 mm × 2.5 mm × 2.5 mm in size, were cut from the foam specimens using a razor blade and were sputter-coated with gold prior to analysis. The samples were investigated in the rising direction at an accelerating voltage of 10 kV. At least 4–5 micrographs were taken for each foam formulation. The cell diameters of the polyurethane foam cells were determined from SEM images using Image J version 1.52a software and the average cell size was calculated using Microsoft Excel. The 10–50 cells were considered for each foam micrograph. Only whole cells were used for measurement.
The densities of the polyurethane foams were determined according to the ASTM D 1622-98 method. Five specimens were cut to have the dimensions of 25 mm ×25 mm × 25 mm (width × length × thickness). After that, these specimens were precisely weighed. The result of the weight (M) divided by volume (V) of the specimens was the density in units of kg/m3
The compressive strength of the foams was evaluated using a Universal Testing Machine (NRI-TS500-30B, Narin Instrument, Thailand), equipped with a 1 kN load cell. The specimens had dimensions of 30 mm × 30 mm × 30 mm (width × length × thickness) and were subjected to compression at a crosshead speed of 3.0 mm/min. The parallel direction of force to the direction of foam growth was applied during measurement. The compressive stress at a 10% deformation of the original thickness was determined. The compressive strength of each foam was determined as the average value of five measurements.
The measurement of water adsorption was performed in accordance with ASTM D2842. The samples were dried at 80°C for 1 h and then weighed. They were then immersed in distilled water to a depth of 1 cm for 24 h. After removal from the water, the samples were held vertically for 10 s to allow for removal of any pendant drops, blotted with dry filter paper for 10 s, and weighed again. The results were obtained as the average of five specimens.
The surface hydrophobicity was assessed through contact angle measurements using the sessile drop method. The measurements were conducted using a Dataphysics Model TC/TPC150, Germany, contact angle instrument. A water drop of 1 μL was deposited onto a flat starch surface made by solution casting method, using a micrometer syringe fitted with a stainless-steel needle. The average value of contact angles were reported.
Results and discussion
Characteristics of sago starch and modified sago starch
The FTIR spectrum of sago starch showed the typical absorption bands associated with a starch backbone (Figure 1(a)). The absorption band at 3304 cm−1 was assigned to hydrogen bonded O-H stretching; 2930 cm−1 was due to C-H stretching; and 1637 cm−1 was attributed to O-H bending vibration. Additionally, a broad absorption band at 994 cm−1, assigned as C-O stretching in C-O-C and C-O-H of the glycosidic ring of sago starch, was observed.
28
In contrast, maleic anhydride-modified starch displayed not only the characteristic absorption peaks of sago starch, but also a C = O absorption peak at 1710 cm−1 (Figure 1(b)).26,29 The O-H stretching peak shifted to a higher wavenumber (3560 cm−1), indicating that free O-H groups were produced after esterification. This confirms that sago starch was modified through esterification and transformed into an esterified product. Typical FTIR spectra of (a) sago starch, (b) esterified sago starch, (c) product prepared from sago starch and p-MDI, and (d) product prepared from esterified sago starch and p-MDI.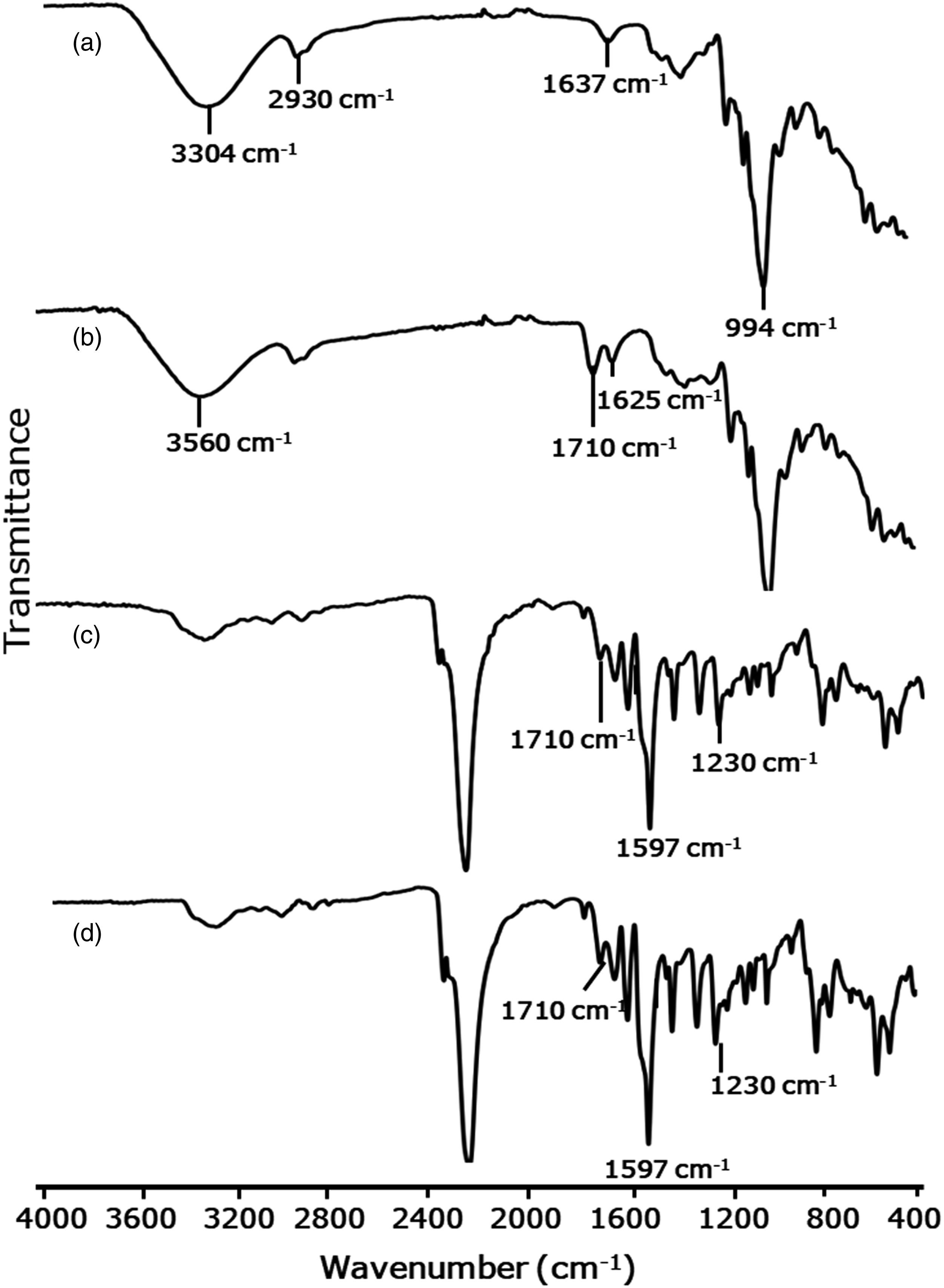
Sago starch and esterified sago starch contain hydroxyl groups which can react with p-MDI. After the reaction with p-MDI, the observable spectral features of the products were related to the dominant reactions, namely reduction of the intensities of bands related to hydroxyl group in the sago starch treated with p-MDI in comparison to the original sago starch (Figure 1(c) and 1(d)). The spectra in Figure 1(c) and 1(d) also show the appearance of carbonyl stretching of urethanes in the spectral region of 1708-1710 cm−1. The other signals are due to the polyurethane-amide linkage at 1597 cm−1 (in plane N-H bending of amides), 1230 cm−1 (C-N stretching, N-H bending of amides).30–32 These features provide evidence that hydroxyl groups on sago starch and esterified sago starch reacted with the isocyanate groups from p-MDI.
The surface morphologies of sago starch and esterified sago starch were studied using a scanning electron microscope (SEM). The SEM images are presented in Figure 2. SEM images of (a) sago starch, and (b) esterified sago starch.
The sago starch granules were shown to be solid circles with a smooth surface and edges in Figure 2(a). In contrast, Figure 2(b) showed that the surface of the esterified starch granules was partially destroyed and became rough. It was noted that the esterification reaction did not deteriorate the entire granule structure of the starch and may be due to the fact that the dry method was performed under anhydrous conditions without starch gelatinization. 26
The particle diameters of esterified sago starch from 10 granules were found to be similar to those of unmodified sago starch after characterizing with a scanning electron microscope as shown in Figure 2(b). The diameter distribution of unmodified sago starch was around 24-35 µm while the diameter of esterified starch was 26-38 µm. This result confirms that esterification reaction mainly happened on the surface of starch particle.
X-ray diffractometer was used to characterize sago starch and esterified starch as shown in Figure 3. XRD patterns of (a) sago starch, and (b) esterified sago starch.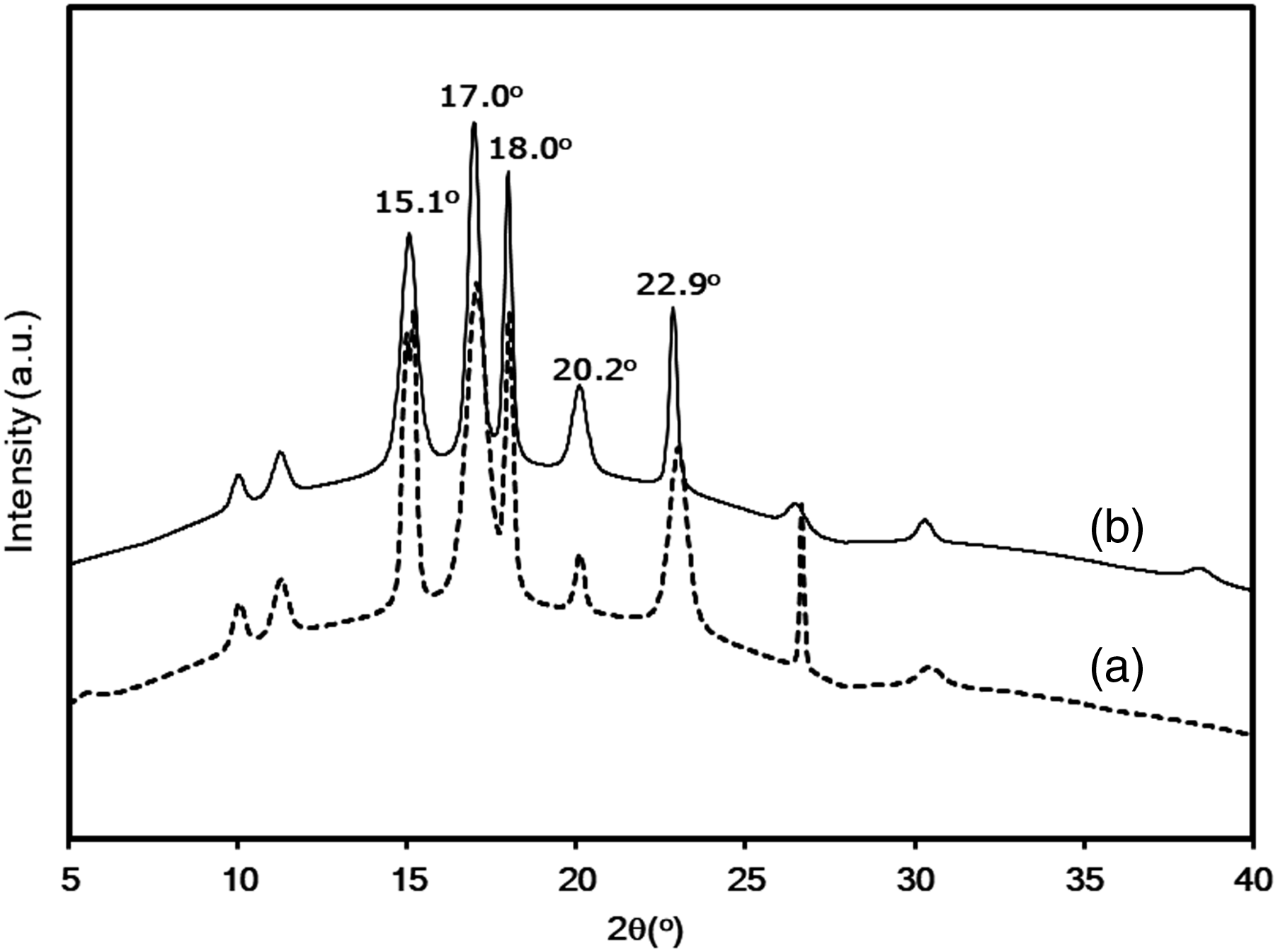
The X-ray diffraction (XRD) results indicated that the sago starch exhibited typical type A crystallization peaks at 2θ values of 15.1°, 17.0°, 18.0°, 20.2° and 22.9°, as previously reported in literature.23,28 The crystallization type of the sago starch remained unchanged after esterification with maleic anhydride, suggesting that the reaction occurred primarily at the surface of the starch granules. The degree of crystallinity of the sago starch was determined to be 66.0%, while the degree of crystallinity of the esterified starch was found to be decreased to 57.7%.
The contact angle measurement, expressed as the tangent of a liquid drop on a solid surface, was used to assess the surface wetting properties of sago starch and esterified sago starch. As shown in Figure 4, the contact angle of water on sago starch was 70° (Figure 4(a)), while it changed to 45.4° on esterified sago starch (Figure 4(b)). The increase in hydrophilicity of the modified starch surface reflects the introduction of ester functional groups during esterification. XRD results indicate a decrease in starch crystallinity, which led to a decrease in hydrogen bonds among starch chains and an increase in the number of free hydroxyl groups, resulting in a more hydrophilic and reactive starch surface with isocyanate groups of p-MDI. Images of the contact angle of (a) sago starch and (b) esterified sago starch.
The rheological properties of polyol premixes is an important parameter determining the overall quality and uniformity of the foam structure.
The results of rheological experiments conducted with polyol-starch mixtures are displayed in Figure 5. The base polyol exhibited Newtonian behavior and the lowest viscosity, as indicated in Figure 5(a). In contrast, the polyol-starch mixtures in Figure 5(b) and 5(c) showed shear thinning behavior, characterized by a rapid decrease in initial viscosities under increasing shear rate, followed by a slower decline at higher shear rates where the starch particles achieved optimal arrangement. This behavior agrees with previous literature reports.7,33,34 Comparing the two types of starches, it was found that the polyol-esterified sago starch mixture had a higher viscosity than the polyol-sago starch mixture. The increased viscosity can likely be attributed to greater swelling of esterified sago starch particles in polyol, compared to the limited swelling of sago starch. This suggests that the interaction between the esterified sago starch and polyol is stronger than that between sago starch and polyol, which is consistent with previous findings.7,35 Rheological behavior of polyol-sago starch mixtures: (a) base polyol, (b) polyol-sago starch mixture, and (c) polyol-esterified sago starch mixture.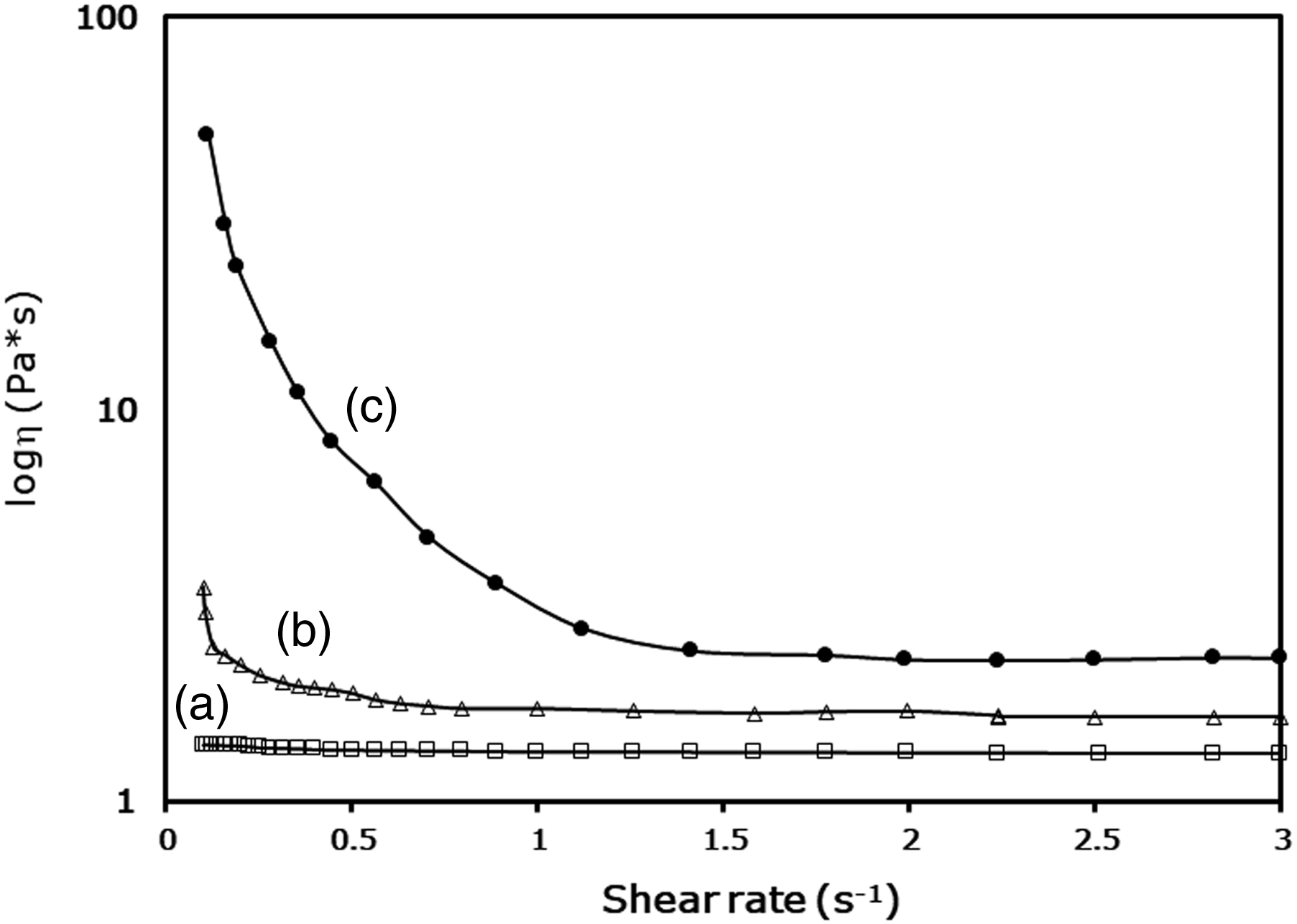
Foaming characteristics
The results of the exothermic temperature profile measurements are presented in Figure 6. This can be attributed to the reaction between the hydroxyl group or water and isocyanate, which generates heat. The maximum exothermic temperature increases with sago starch content indicating the exothermic reaction between hydroxyl group of sago starch and isocyanate (as indicated by FITR spectra in Figure 1). This result is consistent with previous findings regarding the impact of the hydroxyl groups introduced by fillers on temperature rise during foaming process.
7
Exothermic temperature rise during foaming process.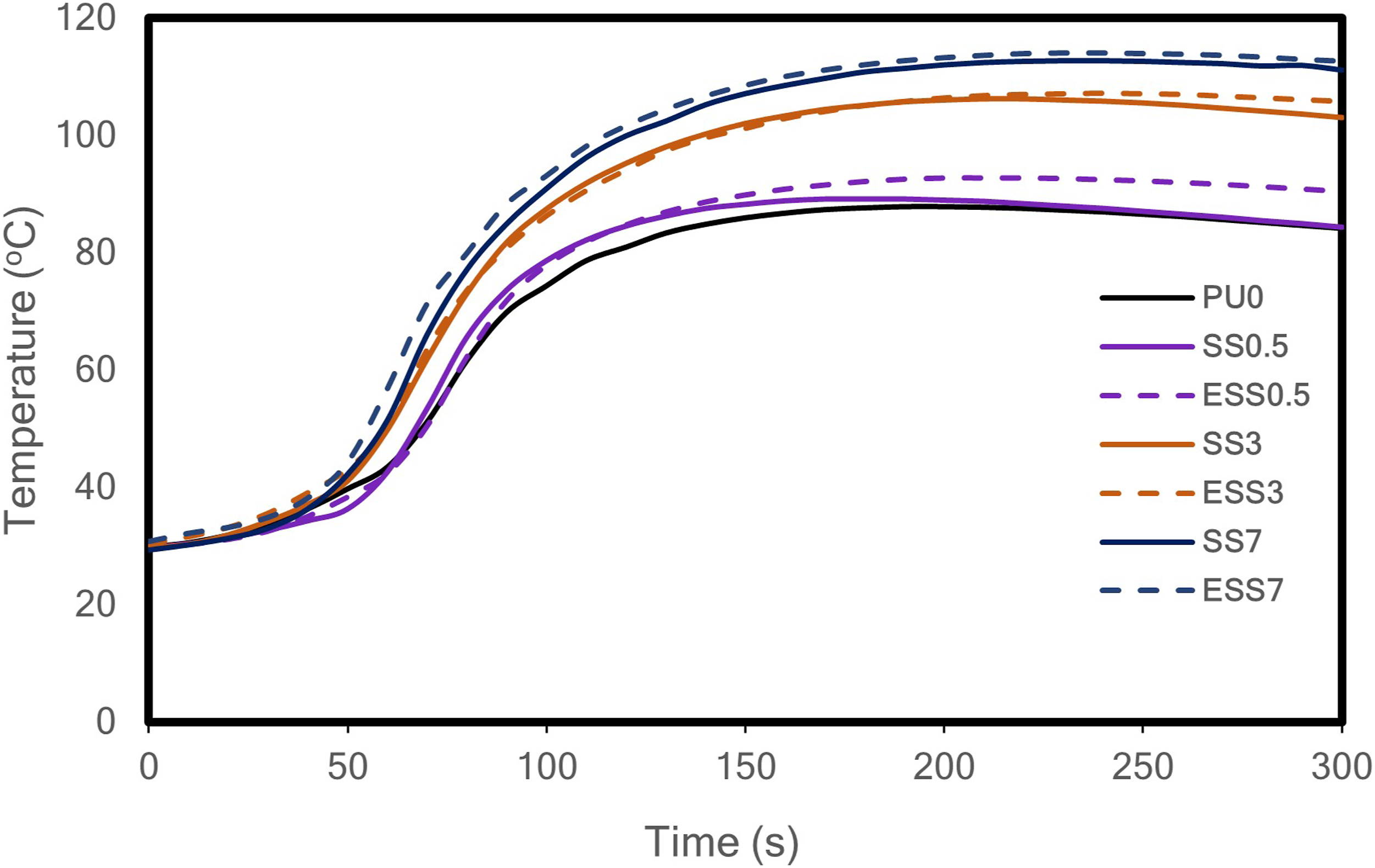
Foam processing times and maximum exothermic temperature.
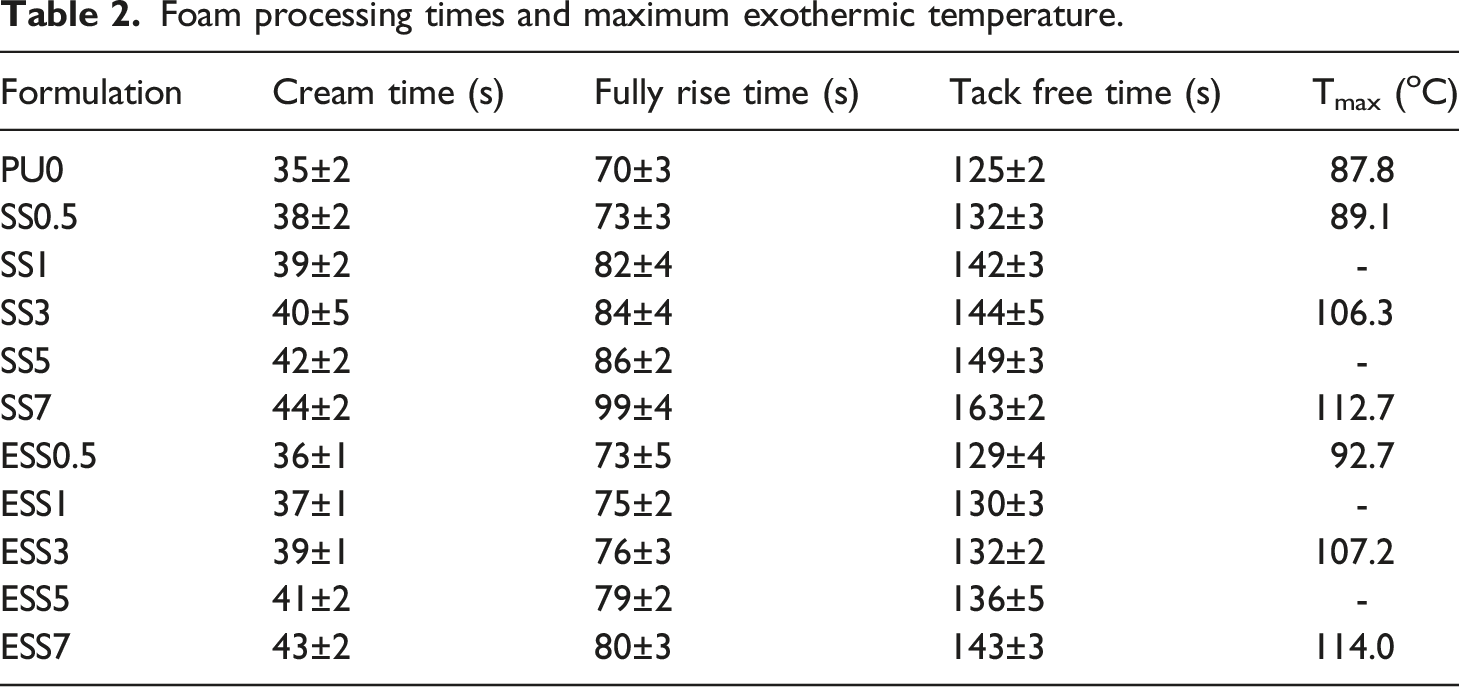
The foaming process was characterized by measuring the characteristic processing times i.e. cream time, fully rise time, and tack free time as reported in Table 2. The results of the experiments show that there was a slight increase in the cream time and full rise time of RPUFs containing starch filler with increasing amount of filler added. This relationship can be attributed to the well-dispersed filler acting as a nucleating agent in the nucleation process, as previously reported in literature,33,36 leading to increased bubble formation and prolonged cream time. The increased viscosity of modified systems, as already discussed, also seems to hinder the further growth of the resulting cells, giving rise to prolonged cream and full rise times, as was also found in the industrial potato protein reinforced rigid polyurethane foam system. 7 However, the processing times were shorter for esterified sago starch filler compared to sago starch filler. This can be due to the decrease in crystallinity of the esterified starch, which may facilitate the diffusion of reactant chains, leading to a slight acceleration of the polymerization reaction, as reported by Grząbka-Zasadzińska and co-workers. 37
Cellular structures
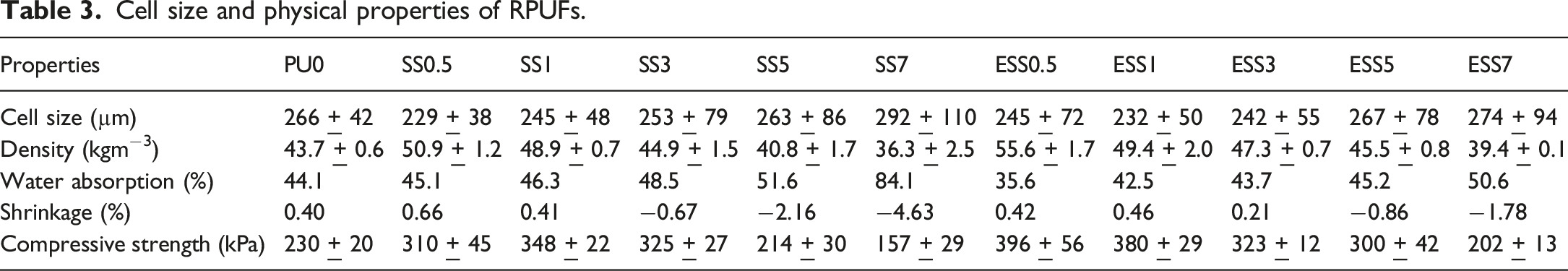
The cross-sectional microstructure of the rigid polyurethane foams (RPUFs) was analyzed using scanning electron microscopy (SEM), as shown in Figure 7. The average cell size was calculated from at least 50 cells, as presented in Table 3. The results indicate that the addition of sago starch and esterified sago starch led to a reduction in cell size and a finer cell structure compared to the control foam (PU0). SEM micrographs of polyurethane foams. Cell size and physical properties of RPUFs.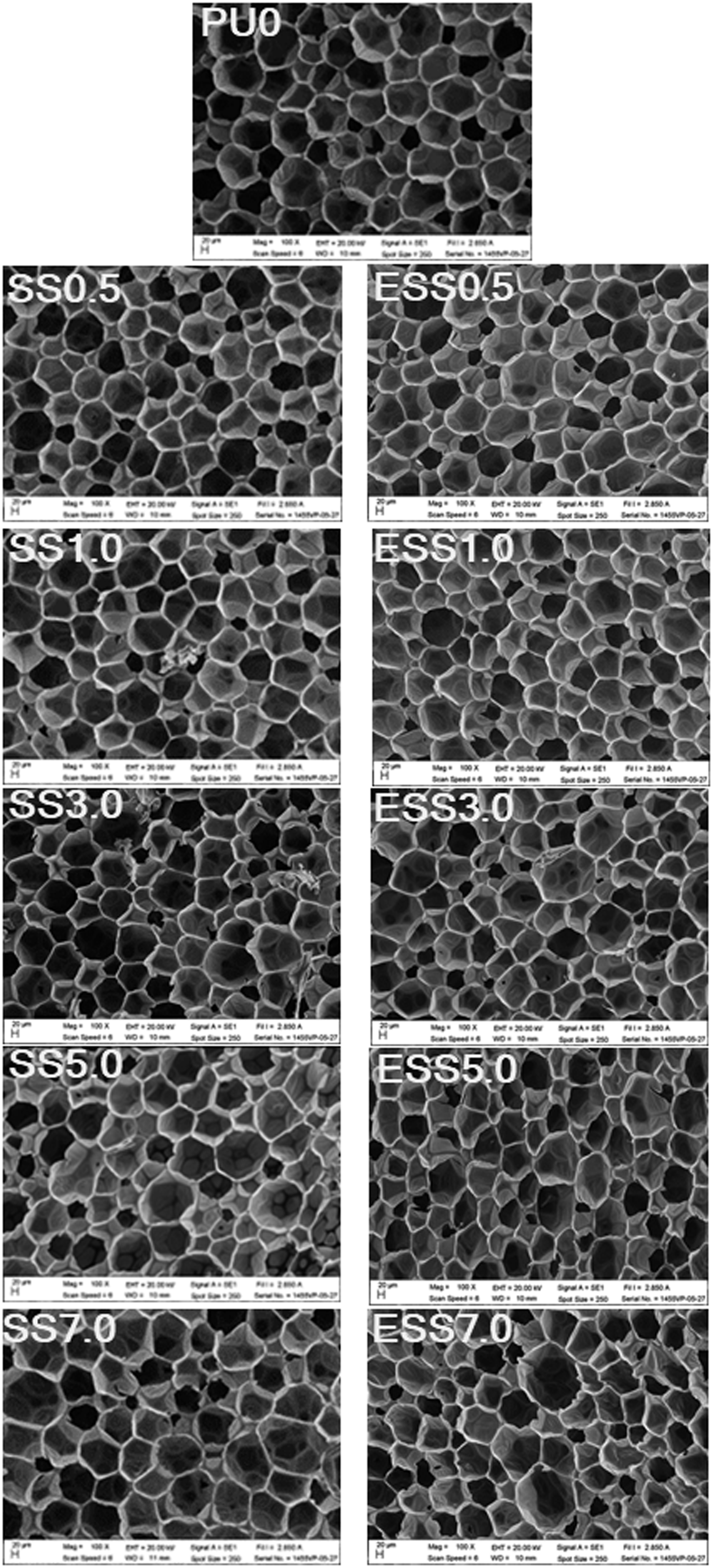
This phenomenon can be attributed to the nucleation effect of starch on gas bubbles. 38 Sago starch and its modified form are believed to act as nucleating agents, enhancing heterogeneous nucleation. Addition of nucleating agent decreases the nucleation energy barrier and increases the number of nuclei. 39 Given that the amount of gas available for foaming is limited, the rapid production of a large number of cells rapidly consumed the blowing gas, resulting in smaller cells with a more uniform distribution. 40 The increase in cell size observed in the RPUFs filled with a higher amount of starch filler can be explained by the concept of coalescence. The large number of cells due to the higher number of nuclei led to the decrease in the thickness of their cell walls. When neighboring bubbles expand, the thin cell walls became stretched that they might rupture and merge randomly to form larger cells. This observation is in accordance with previous findings reported in the literature.41–43
The hydrophilic or hydrophobic nature of the filler plays a crucial role in determining the cell morphology of RPUFs. As described in previous section, the high contact angle value of sago starch indicated its hydrophobic nature. This resulted in poor interfacial adhesion between the sago starch surface and the hydrophilic polyurethane matrix, leading to a higher number of open pores and lower compatibility with the hydrophilic matrix. The rapid cell collapse or cell rupture was further accelerated at higher filler contents of 5.0 and 7.0 wt %. Conversely, a hydrophilic filler's high compatibility with the polyurethane matrix can slow down the film rupturing process during thinning, resulting in a relatively thick film and low likelihood of open pore formation in the polyurethane foams. 43 The presence of open pores was confirmed by the water absorption results reported in the following section.
Physical properties of RPUFs
The composition and microstructure changes of the foams are related to their physical properties. The density of PUF was found to increase slightly with the addition of a lower amount of starch, but then decrease with a further increase in starch content. This increase in density is attributed to the higher density of starch (800-830 kg/m3) 44 compared to neat RPUF. However, the density of RPUF decreased for higher amounts of starch, due to the formation of highly porous cell structure having larger voids (discussed in “Cellular Structures” Section). This result correlated to previous studies which reported the formation of more voids and broken cell structures during the preparation of polyurethane foam at higher loading of fillers.7,33,34,41
The water absorption properties of the porous materials were affected by the ratio of closed and open cells. As demonstrated in Table 3, the water uptake of the RPUFs increased with the increase of starch content, consistent with the increase in the number of open cells in the foam structure.
The cell structure, particularly the quantity of closed cells, played a crucial role in the shrinkage behavior of the RPUF.45,46 The PU0 foam, as well as the RPUFs with sago starch contents up to 1.0 wt% (SS0.5-SS1), showed no significant changes in dimensional stability. However, the application of higher amounts of starch (i.e. SS3-SS7) resulted in a substantial shrinkage of the RPUFs, as depicted in Figure 8. However, the RPUFs filled with esterified starch (ESSs) demonstrated less shrinkage, only showing signs of shrinkage when the esterified starch content reached 5-7 wt% (ESS5-ESS7). Polyurethane foam specimens after preparation.
The shrinkage of the foam was found to increase with higher filler content, as indicated by its structure being less rigid and unable to maintain stability under pressure differentials. The larger cell size and thinner cell walls observed in the foam structure with higher proportions of starch were found to enhance CO2 diffusion, leading to further shrinkage of the final product. 46 Comparing the effect of starch modification, the esterified sago starch gave foams exhibiting less shrinkage than sago starch containing foams. This might be attributed to the better compatibility of the esterified sago starch filler with the polyurethane foam matrix. Sung and Kim 43 explained that PU matrix exhibits hydrophilic characteristic. Poor interfacial adhesion between hydrophobic fillers and the hydrophilic PU matrix can cause rapid cell collapse and this phenomenon is exacerbated with higher filler content.
The RPUFs exhibited a compression behavior as demonstrated in Figure 9, The filled RPUFs displayed higher strength compared to PU0. As shown in Table 3, the compressive strength increased with starch content to an optimal point and then declined. This can be attributed to the reduction in cell size with increasing starch content, leading to larger cell size and increased open cells at high starch loading. The ESS series showed higher compressive strength values compared to the SS series. The addition of 1 wt% sago starch and 0.5 wt% esterified sago starch resulted in a significant improvement of compressive strength (by approximately 50% and 65%, respectively). This improvement is likely due to the favorable chemical affinity and interaction between the starch and polyurethane matrix, leading to better dispersion and efficient load transfer. The compression behavior of RPUFs.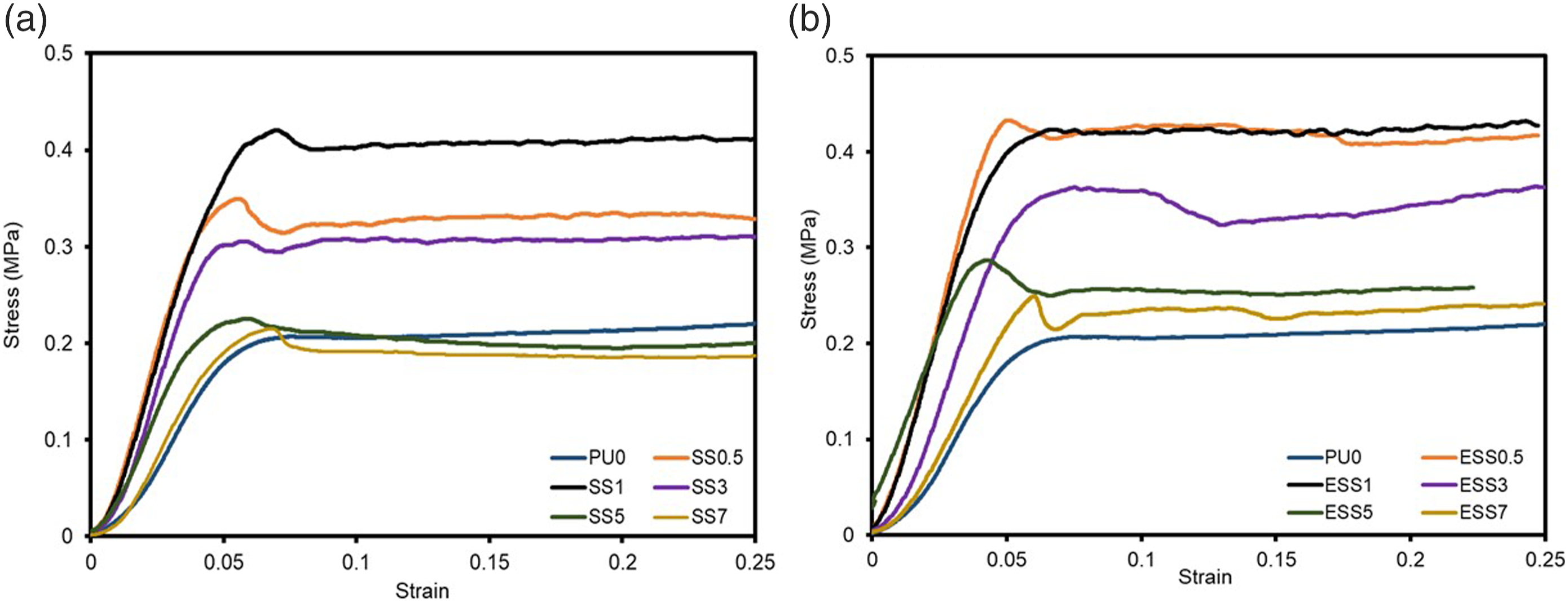
Conclusions
The present study successfully prepared rigid polyurethane foam composites filled with sago starch and esterified sago starch. The esterification process mainly took place on the surface of the starch particle, resulting in lower crystallinity and increased hydrophilicity. This led to shorter processing times and a greater exothermic temperature compared to RPUFs filled with sago starch. The sago starch content had an effect on the cell morphology, where an increase in sago starch content initially resulted in a decrease in cell size, followed by an increase at high loadings of the starch filler. The esterified sago starch showed better compatibility with the polyurethane matrix than the sago starch, which in turn impacted the cellular morphology and physico-mechanical properties of the resulting RPUFs. The water absorption, and volume shrinkage of the RPUFs filled with esterified sago starch were lower than those filled with sago starch, while their density and compressive strength were higher. The optimal results were achieved with 1 wt% sago starch and 0.5 wt% esterified sago starch in the polyol. Overall, the results confirm that the use of esterified sago starch as a filler in rigid polyurethane foam is a viable option for utilizing this natural product as a sustainable resource.
Footnotes
Acknowledgments
The authors would like to thank Bangkok Foam Co.Ltd for providing chemicals used in this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.