
Abstract
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive disorder with a hereditary basis characterized by the degeneration of motor neurons in the anterior horns of the spinal cord and the motor nuclei of the brainstem resulting in muscle weakness and atrophy. 1 Diseases with autosomal recessive transmission occur only in people who have inherited 2 altered copies of a gene.
SMA can be considered as a group of hereditary neuromuscular diseases characterized by muscle thinning and muscle weakness, caused by the progressive degeneration of the nerve cells that receive and transmit motor stimuli (motor neurons) in the spinal cord and brainstem (the part of the brain that connects it with the spinal cord).
They can present heterogeneously both clinically (age of onset, severity of the disease and main site of muscle involvement) and genetically (mode of transmission and genetic defect responsible).
Symptoms may begin in childhood and may include hypotonia, hyporeflexia, difficulty in sucking, swallowing and breathing, failure to reach developmental milestones and very early death in the most severe forms.
Approximately 95% of spinal muscular atrophies (SMA) are caused by deletions of the SMN1 gene.
In a small percentage of patients (5%) at least one of the 2 SMN1 genes has mutations: typically these patients have a deletion on one chromosome and a point mutation on the other chromosome. Around 40 different mutations have been identified to date.
The genetic peculiarity of this disease is that in every individual, in the region of chromosome 5 where the SMN1 gene is present, there is also a gene called SMN2, which has a very similar sequence to SMN1.
This gene differs from SMN1 by a few nucleotides, including one that alters the normal process of transcription of DNA into RNA, leading to the production of a shorter, ineffective protein.
However, 10% to 15% of the proteins produced by the SMN2 gene are the same length as the proteins produced by SMN1 and are fully functional.
This means that in patients with spinal atrophy, the production of full-length SMN protein is not completely absent but is much lower (about 10%) than it normally is.
This allows most of the body’s cells to function properly but not those of the motor neurons in the spinal cord, which degenerate resulting in thinning and weakness of the muscles.
There are 5 types of spinal muscular atrophies 2 :
- Type 0, has prenatal onset and manifests as decreased fetal movement in the last stage of pregnancy. Death occurs within the first 6 months of life and it is caused by respiratory failure.
- Type 1, also known as Werdnig-Hoffmann disease, manifests itself within 6 months of birth and subjects have severe swallowing and breathing difficulties. Death occurs within the first year of life in 95% of cases and within 4 years of life in
- Type 2, also known as Dubowitz’s disease, symptoms appear between 3 and 15 months of age. Children present with profound muscle weakness and are unable to walk or crawl. It is fatal in infancy although the progression may stop spontaneously, with the child characterized by permanent non-progressive weakness that will lead to the use of wheelchairs
- Type 3, or Wohlfart-Kugelberg-Welander disease, appears between 15 months and 19 years of age. The symptoms are similar to Type-1 SMA, but the progression is slower and life expectancy longer. The atrophy tends to affect the lower limbs initially and then affects the arms at a later stage. Life expectancy depends on the appearance of any respiratory difficulties.
- Type 4 appears in adulthood around the age of 30 to 60 years and is characterized by a slow progression of muscle weakness and atrophy. It is very similar in diagnosis to amyotrophic lateral sclerosis and the distinction between the 2 diseases is quite complex.
The most common and severe form (SMA 5q type 1) is the infantile onset form, called “SMA 5q,” as the genetic defect is located on chromosome 5, characterized by the presence of 2 copies of the SMN2 gene; this form occurs in one in 6000 births. 3
The child presents severe hypotonia and proximal hyposthenia and absence of osteotendinous reflexes; there will be a severe delay in reaching motor milestones, not acquiring the ability to control the head or maintain a sitting position.
The diagnosis of spinal muscular atrophy (SMA) is primarily clinical and must be confirmed by genetic testing. The currently most widely used level 1 genetic test is the MLPA (Multiplex Ligation-dependent Probe Amplification) test.
This is a rapid test that can identify deletions or duplications of a gene and thus identify both healthy carriers (carriers of only one altered gene) and individuals with the disease (with both genes altered).
Escaping this test are the 5% of patients who carry a mutation within one of the 2 genes. Since patients with the mutation almost always have one of the 2 SMN1 pairs mutated and the other pair altered by a deletion, the MLPA test only identifies the deletion and it is necessary to determine the DNA sequence of the entire SMN1 gene to identify the mutation.
In addition, the MLPA test allows counting the copy number of SMN2, the gene whose copy number determines the severity of the clinical picture of the disease (SMA type 1, type 2, type 3, and type 4). 4
The speed of disease progression is related to the age of onset and is usually rapid. In the final stages, the child presents immobile, with the upper limbs in adduction and the lower limbs in a frog-like posture. Life expectancy is less than 2 years, which is why the 3 recent therapies approved in the last 7 years are crucial in improving the patient’s condition and prolonging life expectancy.
- Nusinersen, administered lumbar in 4 doses post-diagnosis and with a maintenance dose once every 4 months. It acts as an antisense oligonucleotide allowing the synthesis of full- length functioning SMN protein.
- Onasemnogene abeparvovec, gene therapy administered via single dose intravenous infusion. It introduces a functional copy of the SMN1 gene by promoting survival and functionality of transduced motor neurons.
- Risdiplam, administered orally once a day. It increases production and sustains levels of functional SMN protein.
Onasemnogene abeparvovec in Europe can be prescribed with a hospital prescription: medicines subject to a restrictive medical prescription that can only be used in hospital certified by the manufacturer or similar establishment.
The following study looked at patients treated with onasemnogene abeparvovec, a drug whose active ingredient expresses the human motor neuron survival protein (SMN1).
It is a vector based on a recombinant adeno-associated serotype-9 virus (AAV9), which is unable to replicate, containing cDNA (copy DNA) for the human SMN gene. This gene therapy will introduce a functional copy of SMN1 into the transduced cells to provide an alternative expression of the SMN protein in motor neurons, thus promoting their survival and functionality.
The patients who underwent the infusion process in our hospital (Fondazione Policlinico Universitario Agostino Gemelli IRCCS - Rome) all had an established diagnosis of SMA 5q type 1, with a biallelic mutation of the SMN1 gene and 2 or 3 copies of SMN2.
The aim of the study was to collect data from the re-evaluations of subjects treated with onasemnogene abeparvovec, and to assess their CHOP-INTEND score to check whether the patient’s health status had improved and whether there were any serious adverse effects following treatment.
These results were then compared with those of the registration study (CL-303) reported in the Summary of product characteristics (SPC). SPC is a document describing the properties and the officially approved conditions of use of a medicine. Summaries of product characteristics form the basis of information for healthcare professionals on how to use the medicine safely and effectively.
Methods
In this project, data were extracted through the re-evaluations entered in individual patient records within the AIFA monitoring registers.
To the 16th February 2023, there were 11 patients treated, but only 8 were taken into account as 3 of these are still undergoing post-infusion monitoring. Of the 8 patients who presented complete data, the scores considered were eligibility, re-evaluation 1, re-evaluation 2, re-evaluation 3 and end of treatment.
There were 3 re-evaluations per individual patient approximately 1, 3, and 6 months after onasemnogene abeparvovec was administered, as described in AIFA guidelines.
The data include:
- Patient details: patient’s weight, date of disease onset, age at disease diagnosis and at individual re-evaluations, date of patient status assessment.
- Diagnosis: SMA 5q detection, SMN2 copy number, type of SMA, presence of symptoms in the patient.
- Patient symptoms: head control, ability to sit, ability to stand, ability to walk, ability to feed, presence of assisted ventilation.
- CHOP-INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders)
- HINE assessment (Hammersmith Infant Neurological Examination)
Of the 11 patients in the analysis set, all the 11 patients (100%) were Caucasian and there are no other ethnicities in our study population.
Sixty-three percent of the treated patients (5/8) showed dysphagia as a comorbidity and 100% (8/8) had Werdnig-Hoffman as a type of SMA.
The patients in the study were all patients treated within the Fondazione Policlinico Gemelli (FPG) in Rome and no eligibility or inclusion criteria based on gender were applied.
Results
Patient data were collected in the following table (Table 1):
Treated Patients’ Data. The minimum and maximum values found in the collected data are shown in brackets.

At regular intervals of 1, 3, and 6 months, re-evaluations of the patients’ clinical conditions were carried out and, through the CHOP-INTEND score, their health status was scored to assess any progress and improvements.
The scores obtained are shown in the Table 2 below:
CHOP-INTEND Scores of Fondazione Policlinico Gemelli Patients.
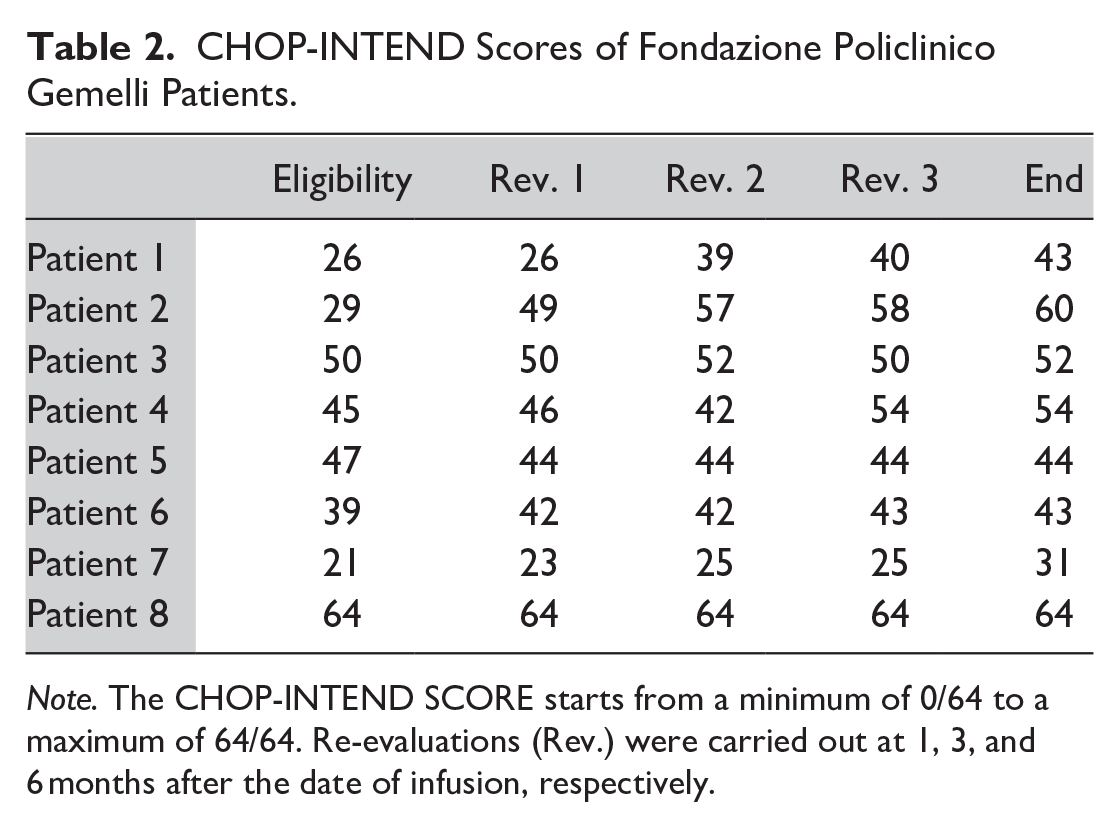
Note. The CHOP-INTEND SCORE starts from a minimum of 0/64 to a maximum of 64/64. Re-evaluations (Rev.) were carried out at 1, 3, and 6 months after the date of infusion, respectively.
Once these data had been extracted, the CHOP-INTEND scores of the individual patients were graphically represented and used as a parameter to assess the development of the health status of the treated subjects (maximum score 64/64). These values were then compared with the data reported in the drug registration study CL-303. 5
The 8 patients examined all had an established diagnosis of SMA 5q with a biallelic mutation in the SMN gene and a SMN gene copy number varying between 2 and 3.
The age at diagnosis ranged between 0 and 15 months and monitoring was also carried out up to 38 months of age.
The CHOP-INTEND scores of the treated FPG patients were thus plotted in a line graph as visible in Figure 1.
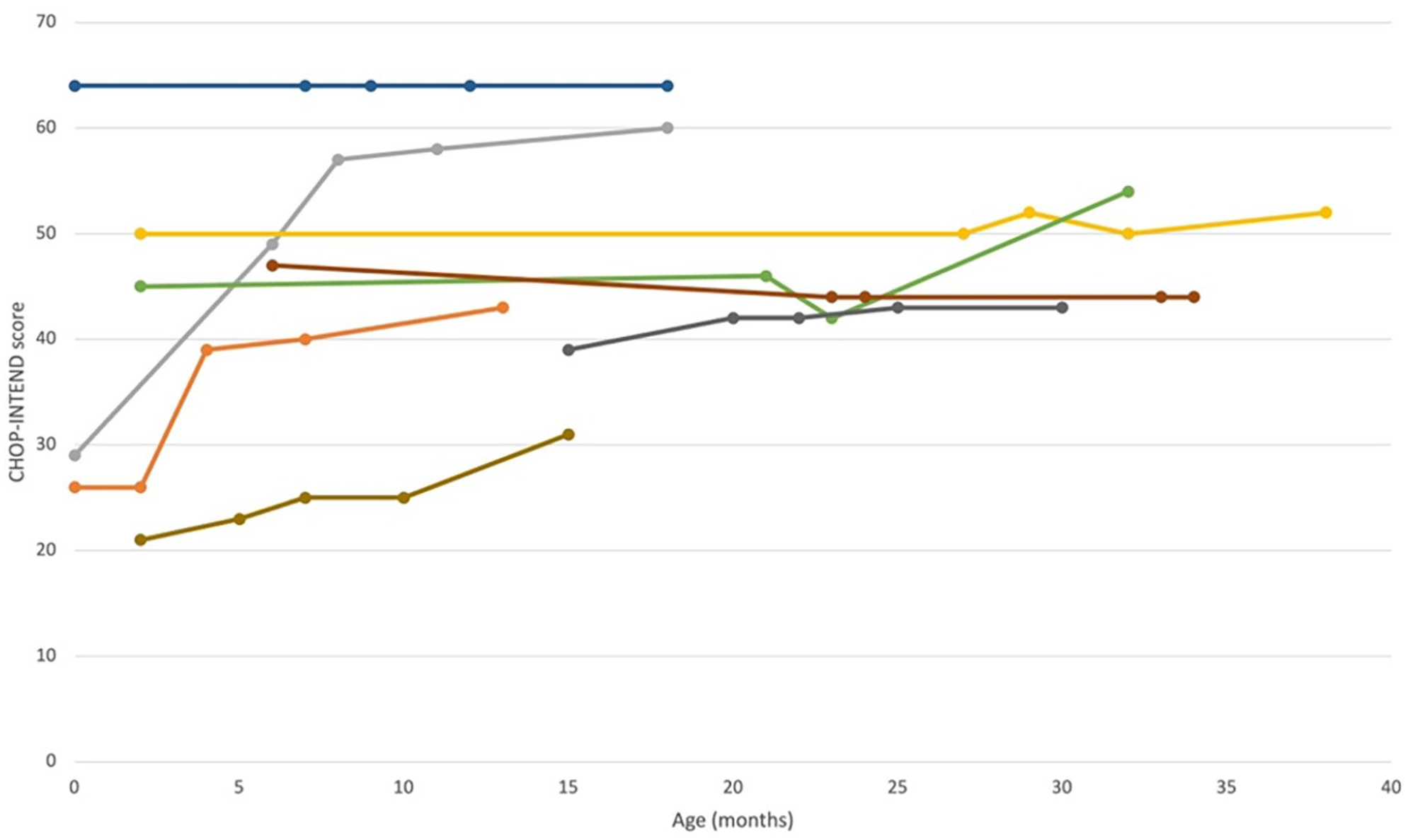
Graphical representation of CHOP-INTEND scores of treated Fondazione Policlinico Gemelli patients. Each line represents a patient undergoing treatment with onasemnogene abeparvovec at Policlinico Gemelli.
Of the scores obtained, the Δ between the score of each re-evaluation and the initial score was calculated so that the subjects with the highest score increase could be observed more quickly (Figure 2).
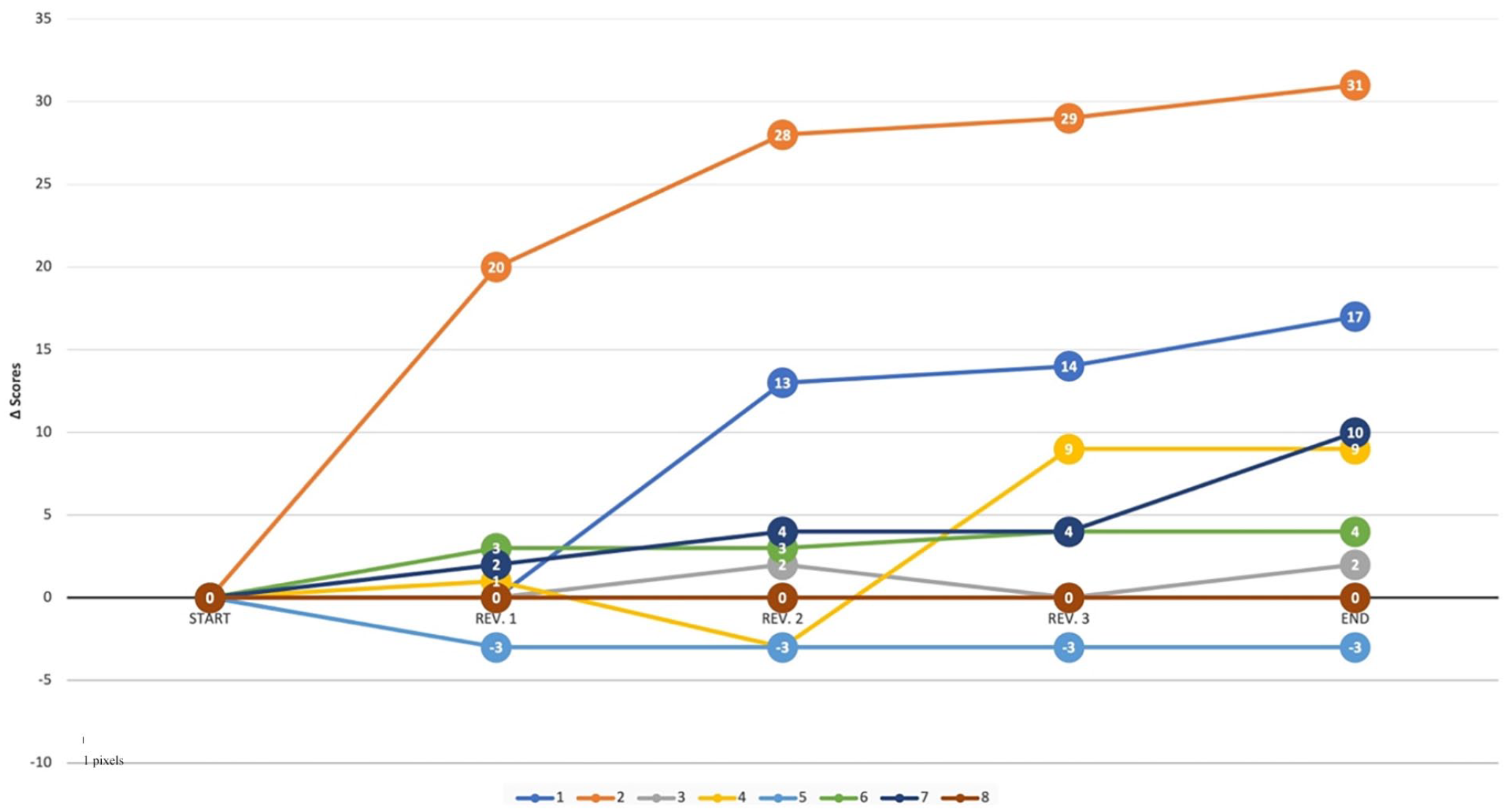
Δ Scores representation of CHOP-INTEND scores of treated Fondazione Policlinico Gemelli patients. Each line represents a patient and each point has a value equal to the difference between the score recorded during each individual re-evaluation and the score recorded by the patient before undergoing treatment. All but one of the patients show a positive delta score, synonymous with an improvement in their clinical state.
These values were then placed within a graph that also included the scores from the CL-303 registration study, as depicted in Figure 3: in blue are the patients examined in the CL-303 study while in red are the patients treated at Fondazione Policlinico Gemelli Hospital.
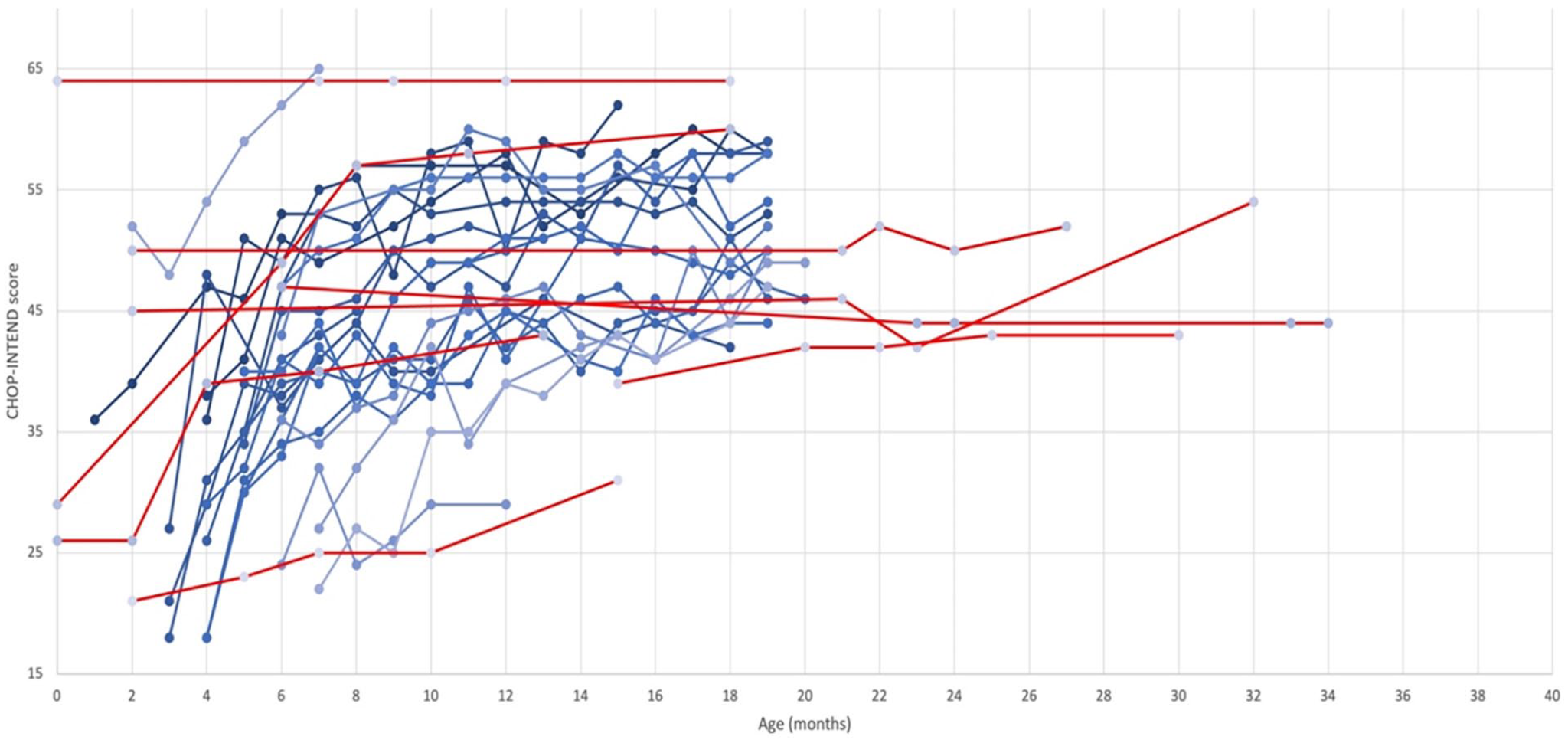
Comparison of CHOP-INTEND scores from the CL-303 study (blue) and Fondazione Policlinico Gemelli patients (red). Our results (red lines) confirm the positive post-infusion trend in the increase of the score recorded at each re-evaluation as visible from the overlap with the data reported in the registration study (blue lines). Each point represents the month of age of the child in which the revaluation was carried out. Color version of the figure is available online.
Clinically significant responses were those that led to an increase in the CHOP- INTEND score of at least 4 points. 6
An evaluation of the scores obtained shows that an increase of ≥4 points in the CHOP-INTEND score was achieved in 5 out of 8 patients; one of the 8 patients, moreover, started with a score of 64 and managed to maintain this score even at the end of all re-evaluation processes.
Observing our data, of the 8 patients treated, one, who initially received non-invasive assisted ventilation, no longer required ventilatory support at the end of the treatment.
Three out of 8 patients were able to maintain head control at the end of the treatment and 5/8 were able to maintain a sitting position for at least 30 seconds without any support; only one patient was able to ambulate without any help at the end of the monitoring period.
Of the patients treated with onasemnogene abeparvovec, 6 out of 8 had previously been treated with nusinersen for a range of time between 2 and 21 months, before switching to gene therapy.
Discussion
Data Analysis and ADRs
A series of data from various clinical trials on patients with SMA-1 and SMA-2 show that the efficacy of therapy increases if carried out immediately after or before the onset of symptoms. Therefore, prenatal screening is essential in order to verify the health status of the child and ensure the best possible treatment as soon as possible after birth.
Given the high cost of onasemnogene abeparvovec therapy, a comparison can be made between the costs of this therapy and nusinersen therapy: €420 000 of nusinersen for the first year for 6 injections followed by €210 000 per year for 3 injections per year as opposed to €2.1 million for the single administration of onasemnogene abeparvovec; in the case of continued therapy with nusinersen, the cost of such therapy from year 8 onward will be the same as those with onasemnogene abeparvovec.
Our study was an analysis of the CHOP-INTEND scores of patients treated with onasemnogene abeparvovec within the Fondazione Policlinico Universitario Agostino Gemelli IRCCS.
The CHOP-INTEND score is a tool developed by the Children’s Hospital of Philadelphia to assess whether a given treatment leads to a change in the course of the disease in children with SMA-1. There are 16 parameters taken into account and these are given a score ranging from 1 to 4 for a total score of 64.
A change in score between eligibility and end of treatment ≥4 was considered clinically significant and synonymous with an improvement in the patient’s clinical condition.
The main adverse reactions reported in SCP following infusion mainly concern immunogenicity phenomena such as immune-mediated hepatotoxicity, resulting in increased Alanine aminotransferase (ALT) and Aspartate aminotransferase (AST) values, as manifested by some cases of death from liver damage occurring in recent months following the infusion.
The underlying mechanism is likely related to an innate and/or adaptive immune response to the vector. A prophylactic corticosteroid regimen and monitoring of liver function at baseline and regularly for at least 3 months after onasemnogene abeparvovec infusion are therefore recommended. This includes weekly monitoring for the first month, and during the entire corticosteroid tapering period, followed by every 2 weeks for another month, and at other times if clinically indicated.
Recently, 2 fatal cases of acute liver failure have been reported in patients with SMA treated with onasemnogene abeparvovec, at 4 and 28 months of age respectively. Common clinical characteristics of these 2 cases are summarized below:
The initial manifestation of liver injury was asymptomatic elevation of liver aminotransferases within the first 1 to 2 weeks post onasemnogene abeparvovec infusion, which was treated with an increased prednisolone dose.
The clinical presentation of hepatotoxicity included vomiting, weakness and a second elevation of liver aminotransferases. This was seen between 5 and 6 weeks post onasemnogene abeparvovec infusion, and approximately 1 to 2 weeks after the initiation of the prednisolone taper.
Rapid deterioration in liver function, and progression to hepatic encephalopathy and multi-organ failure followed. Death occurred 6 to 7 weeks after the onasemnogene abeparvovec infusion, during the period of corticosteroid dose tapering.
There are also thrombocytopenia phenomena, related to thrombotic microangiopathy (TMA) cases.
TMA is diagnosed by the presence of thrombocytopenia, hemolytic anemia, and acute kidney injury, and occurs due to dysregulation and/or excessive activation of the alternative complement pathway. Its etiology can be genetic or acquired. TMA is treatable and can resolve with timely and proper interventions. It is important to have increased awareness of TMA for patients receiving onasemnogene abeparvovec. In total, 5 confirmed cases of TMA in patients aged 4 to 23 months have so far been reported after treatment with onasemnogene abeparvovec, among approximately 800 treated patients. In these 5 cases, TMA developed within 6 to 11 days after onasemnogene abeparvovec infusion. The presenting features included vomiting, hypertension, oliguria/anuria, and/or edema.
In the acute phase, all patients responded well to medical interventions including plasmapheresis, systemic corticosteroids, transfusions and supportive care. Two patients underwent renal replacement therapy (hemodialysis or haemofiltration). Unfortunately one patient who required renal replacement therapy (haemofiltration) died 6 weeks after the event.
Therapy should not be initiated in the presence of concomitant infections as this could lead to a worsening of the patient’s clinical condition.
In our case, the number of SMA patients who experienced adverse reactions following the infusion was 0. One patient undergoing treatment died from causes unrelated to the drug. Looking at the data reported on the EudraVigilance portal, as of 18 February 2023, there were 525 ADRs related to onasemnogene abeparvovec treatment across Europe.
Of the 546 adverse reactions that occurred, 40 were recorded in Italy, making up 7% of the cases. The largest number of reported adverse reactions concerned gastrointestinal problems and general disorders, as shown in Table 3.
Onasemnogene Abeparvovec ADRs Divided by Groups.
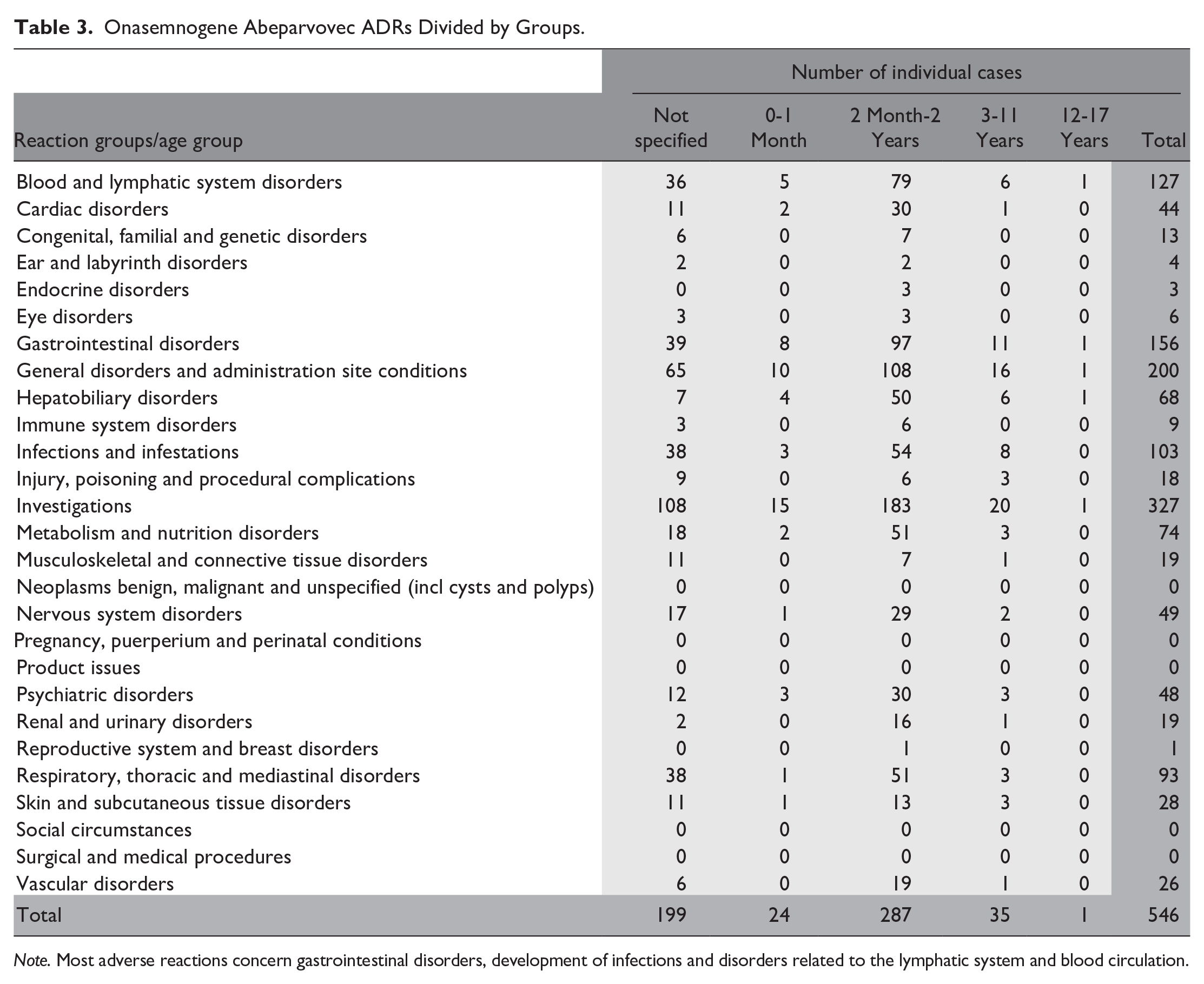
Note. Most adverse reactions concern gastrointestinal disorders, development of infections and disorders related to the lymphatic system and blood circulation.
Other types of ADRs tend to be an increased number of infections, respiratory problems and hepatobiliary disorders.
Ordering, Receiving, Storage, Preparation and Handling Procedures Within the Fondazione Policlinico Universitario Agostino Gemelli IRCCS
Order, reception, and control
When the order is placed, the quantity of onasemnogene abeparvovec requested from the manufacturer is slightly higher than the amount needed for the individual patient because, as the dosage is related to the patient’s weight (1 × 1014 vector genomes per kilogram vg/kg) and as we are dealing with new-born subjects, the weight changes daily; the names of the pharmacists authorized to receive and handle the drug will also be indicated when the order is placed.
The vials of onasemnogene abeparvovec, after being transported in temperature-controlled containers (≤−60°C), must be stored at a temperature between 2°C and 8°C inside a refrigerator and used within 14 days of receipt, not refreezing and shaking the vials. The package contains a data logger capable of recording the temperature and any changes in it during the entire shipping and delivery process. Before releasing the batch, the pharmacist will check that no interruption of the cold chain has occurred during the delivery process.
Before use, the vials should be thawed either at room temperature in about 4 hours or after 12 hours in a refrigerator.
At the end of thawing, the absence of particulate matter or color changes inside the vial will be checked.
Set-up, administration, and handling
Prior to administration, the patient should undergo a series of tests (liver functioning, AAV9 antibody titer, creatinine levels, troponin I levels, complete blood count). Monitoring of the following parameters should be rechecked after the infusion.
Twenty-four hours prior to the infusion, systemic corticosteroid administration should be continued post-infusion.
The infusion generally takes place the day after the vials are received, and proper management of the entire process requires synchronization between ward doctors and nurses and UFA (Antiblastic Drugs Unit, Italian acronym UFA) pharmacists and nurses to ensure efficacy and safety of the treatment.
In order to ensure the infusion by 12:00 noon, the drug preparation is scheduled between 10:00 and 11:00 A.M., blocking the activities and allowing 2 nurses to proceed with the set-up in the sterile galenical laboratory, after obtaining a final confirmation from the department, which in the meantime will have transferred the patient to the PICU (Pediatric Intensive Care Unit).
Once the set-up process is completed, the therapy will then be delivered to the PICU in order to proceed with the infusion. The drug will be administered via a slow infusion over 60 minutes (not as a bolus or rapid intravenous administration) and at the end the lines will be flushed with saline solution.
Procedures must be carried out under sterile conditions, wearing PPE such as gloves, safety goggles and a lab coat. In the event of drug spills, surfaces must be cleaned with absorbent gauze and disinfected using bleach solutions first followed by alcohol. Any waste resulting from the preparation of the drug should be disposed of as biological waste, storing it in a double bag. In case of contact with the skin, clean the affected area thoroughly with soap and water for at least 15 minutes while in case of eye contact use water only.
Conclusions
From the data obtained, albeit in a small sample, in our case treatment with onasemnogene abeparvovec was safe and improved the clinical condition of the treated subjects.
The future goal is to collect data from subjects treated with onasemnogene abeparvovec throughout the countries to increase the observed sample and obtain further confirmation of the data.
The limitations of the study may be related to:
- small number of patients examined;
- hospital catchment area of subjects undergoing treatment with onasemnogene abeparvovec covering the center-south of the Italian peninsula.
It became clear that treatment with onasemnogene abeparvovec is an excellent therapeutic option for SMA patients, both in terms of treatment efficacy and high compliance related to the way of administration (one-time injection), compared to other therapies such as nusinersen (1 injection every 4 months).
The effectiveness of this treatment will also have to be evaluated in patients where it will be administered at a later stage of their lives or in a more critical condition of the disease.
There remain issues related to the cost of the drug and uncertainties related to both the efficacy and safety profile, particularly the potential risk of cardiac, dorsal root ganglion toxicity, or long-term carcinogenicity that will be further characterized in ongoing PAES studies.
Footnotes
Author Contributions
MF conceived the study, provided and analyzed the data, wrote the manuscript. DT conceived the study, provided and analyzed the data, participated in the writing of the manuscript and critically reviewed it. LDC provided and analyzed the data, critically reviewed the manuscript. AS provided and analyzed the data, critically reviewed the manuscript. RC critically reviewed the manuscript. MP critically reviewed the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval
Ethical approval was not required for this study.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.