
Abstract
Objectives:
Large-scale genetic analysis of common variation in schizophrenia has been a powerful approach to understanding this complex but highly heritable psychotic disorder. To further investigate loci, genes and pathways associated more specifically in the well-characterized Australian Schizophrenia Research Bank cohort, we applied genome-wide single-nucleotide polymorphism analysis in these three annotation categories.
Methods:
We performed a case–control genome-wide association study in 429 schizophrenia samples and 255 controls. Post-genome-wide association study analyses were then integrated with genomic annotations to explore the enrichment of variation at the gene and pathway level. We also examine candidate single-nucleotide polymorphisms with potential function within expression quantitative trait loci and investigate overall enrichment of variation within tissue-specific functional regulatory domains of the genome.
Results:
The strongest finding (p = 2.01 × 10−6, odds ratio = 1.82, 95% confidence interval = [1.42, 2.33]) in genome-wide association study was with rs10252923 at 7q21.13, downstream of FZD1 (frizzled class receptor 1). While this did not stand alone after correction, the involvement of FZD1 was supported by gene-based analysis, which exceeded the threshold for genome-wide significance (p = 2.78 × 10−6).
Conclusion:
The identification of FZD1, as an independent association signal at the gene level, supports the hypothesis that the Wnt signalling pathway is altered in the pathogenesis of schizophrenia and may be an important target for therapeutic development.
Introduction
Schizophrenia is a severe psychotic disorder associated with substantial morbidity and mortality, as well as great personal and societal costs (Knapp et al., 2004; Leucht et al., 2007; Mathers, 2008; Saha et al., 2007). The aetiology of schizophrenia is complex and multi-factorial, reflecting the combined influence of genetic, environmental and social factors (Gejman et al., 2010; Lavretsky, 2008; Siever and Davis, 2004). While our understanding of the molecular basis of schizophrenia remains incomplete, several theories have been advanced, focusing on dysfunction in neurotransmitter systems, including dopamine, glutamate, serotonin and gamma-aminobutyric acid (GABA) (Patel et al., 2014). Although antipsychotic agents targeting these systems have been developed for the treatment of schizophrenia, current pharmaceuticals are not a complete solution. Between 10% and 30% of patients experience little or no symptomatic improvement, with up to an additional 30% of patients showing partial response to treatments and a 30% risk of relapse per year (Lehman et al., 2004). While drug development for psychotic disorders has been relatively stagnant in recent years, new insight from more comprehensive analysis of the pathophysiology underlying these disorders may provide promising paths to new treatment approaches.
Epidemiological studies have identified the importance of genetics in schizophrenia, with heritability estimates ranging from 64% to 81% (Lichtenstein et al., 2009; Sullivan et al., 2003). Genome-wide association studies (GWASs) have greatly advanced our understanding of the genetic architecture of many psychiatric disorders (Sullivan et al., 2012). The application of this approach in schizophrenia has been particularly successful due to collaborative efforts that have the power to detect small to moderate contributions from many genes. These analyses have also highlighted the vast genetic complexity of the disorder which typically involves the aggregated effect of many variants. Published studies have discovered more than 100 novel genetic loci associated with schizophrenia using GWAS, and in particular, genetic evidence for specific variants emerged in the recent and largest GWAS meta-analysis by the Schizophrenia Working Group of the Psychiatric Genomics Consortium (PGC2) (Ikeda et al., 2011; International Schizophrenia Consortium et al., 2009; Ripke et al., 2013; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Trans-ancestry meta-analysis was further performed in PGC2 samples and a Chinese population, with additional loci reported (Li et al., 2017).
While genome-wide studies have yielded many loci with potentially causative candidate genes, to further understand the biology of these associations and their heterogeneity, we need higher resolution analyses of both the genome and phenome of the disorder. With complex patterns of linkage disequilibrium (LD), the application of regional GWAS and functional genomics should ultimately establish a more refined picture of the genes and networks that contribute to the pathophysiology. In the current study, we conducted a GWAS in an Australian population from the Australian Schizophrenia Research Bank (ASRB), to investigate common genetic variations associated with susceptibility for the disorder.
Materials and methods
Participants in this study
Participants included 786 individuals (476 cases and 310 controls) sourced from the ASRB (Loughland et al., 2010). Diagnosis of schizophrenia was confirmed using the OPCRIT algorithm applied to the Diagnostic Interview for Psychosis (DIP), according to the International Classification of Diseases, 10th Revision (ICD-10). Controls were healthy volunteers with an absence of psychiatric disorders and no history of psychosis in first-degree relatives. The study was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the Hunter New England Human Research Ethics Committee (HNEHREC) with written informed consent obtained from all participants.
Genotyping, quality control and imputation
Genomic DNA samples were extracted from peripheral blood mononuclear cells and genotyped using the Illumina Human610-Quad BeadChip (620,901 common single-nucleotide polymorphism [SNP] markers). After microarray scanning, the raw image intensity data were processed by the genotype module in GenomeStudio (Illumina). Variant calling was performed using GenCall with a minimum no-call threshold of 0.15 in accordance with microarray manufacturer’s guidelines (Illumina). In other word, clusters with a GenCall score less than 0.15 are not assigned genotypes. Problematic samples with call rates below 95% were also excluded. Rigorous quality control was performed, including checking for sex discordance and removing samples with missingness greater than 10%. To evaluate cryptic relatedness, identity-by-descent state (IBS) analysis was performed to exclude closely related samples. Subsequently, population stratification by principal component analysis (PCA) was examined using EIGENSTRAT (Price et al., 2006), comparing the distribution of principal components for the studied samples and five major reference populations obtained from the 1000 Genomes Project Phase III integrated release version 5 (Europeans, Americans, Africans, East Asians and South Asians, Supplementary Figure 1A). Autosomal SNPs with minor allele frequencies (MAFs) >0.01, missing call rates <0.01 and Hardy–Weinberg equilibrium (HWE) exact test p values > 1.0 × 10−6 were used for PCA analysis. They were further pruned by removing one SNP from a pair where pairwise LD exceeded 0.2 within a 50-SNP window before moving on 5 SNPs. As expected, the majority of the Australian population clustered with Europeans. A small proportion of the population showing Asian or African ancestry was excluded to retain subjects with European ancestry, by removing 62 individuals who deviated from the mean of 1000 Genomes Project collective European populations by >6 times their standard deviation (SD) on principal component 1 (PC1) and PC2 (Mishra et al., 2012) (Supplementary Figure 1A). PCA was further performed only in the retained cases and controls to evaluate population substructure (Supplementary Figure 1B), in which the first two eigenvectors were chosen as covariates for the association analysis to correct for the substructure. Quality control procedures excluded non-autosomal SNPs and those with call rates less than 99% or MAFs less than 1%, or SNPs that deviated from HWE with p <1.0 × 10−6 in the control group. Genotype imputation was performed using the 1000 Genomes Project Phase III integrated release version 5 for European population as reference. Samples were pre-phased using SHAPEIT2 (Delaneau et al., 2011), and the data were then imputed with Minimac3 using the reference panel (Das et al., 2016). Stringent imputation quality control was subsequently applied by excluding from further analysis SNPs with RSQR < 0.9, MAFs < 0.01 and HWE deviation test statistics < 1.0 × 10−6. Our final dataset comprised 6,473,404 SNPs for 429 schizophrenia cases (287 males and 142 females) and 255 controls (112 males and 143 females).
Association analysis
Logistic regression analysis was performed using PLINK 1.9 (Chang et al., 2015) fitting an additive model, while adjusted for three eigenvectors and gender. A quantile–quantile plot and the genomic inflation factor (λGC) were generated to examine population substructure (Supplementary Figure 2). The λGC was 1.00 in our study, indicating the impact of population structure was negligible. Conditional analysis was subsequently carried out for detection of secondary association signals
Gene, pathway and expression quantitative trait locus analyses
To explore potential genes and pathways associated with schizophrenia in the GWAS dataset, we applied gene-based and pathway enrichment analyses using MAGMA (De Leeuw et al., 2015) and MAGENTA (Segre et al., 2010), respectively. The gene-based analysis in MAGMA uses a multiple regression approach to detect multi-marker effects, taking into account gene size, the number of SNPs in a gene and LD between SNPs in a gene. For gene-based analysis, all 19,427 protein-coding genes from the National Center for Biotechnology Information (NCBI) 37.3 gene definitions were used as the basis for the analysis in MAGMA. After SNP annotation, using a 10-kb upstream and 10-kb downstream window to include gene regulatory regions, there were 17,950 genes implicated by at least one schizophrenia-associated SNP. Gene association tests were performed, and a stringent Bonferroni correction was applied to account for multiple testing, setting the significance threshold at 2.79 × 10−6 (0.05/17950).
Pathway analysis was performed by combining SNP p values into gene scores, while also correcting for confounding factors, such as gene size, SNP density and LD patterns. MAGENTA pathway analysis was performed using default settings, and an empirical pathway enrichment p value was generated for each pathway derived from different databases (Gene Ontology, KEGG, PANTHER, Biocarta and Reactome). The MAGENTA built-in false discovery rate (FDR) method was applied to correct for multiple testing, and individual pathways that reached FDR less than 0.05 were deemed significantly associated with schizophrenia susceptibility. Finally, to investigate the functional mechanisms underlying the genetic associations, cis-expression quantitative trait locus (eQTL) analysis was applied. Expression data for specific tissues obtained from the Genotype-Tissue Expression (GTEx) portal (Data release V7) were used to evaluate by linear regressions whether the identified variants influence transcript levels of genes as cis-effect regulators.
Enrichment of GWAS summary statistics in functional and regulatory elements
The integrative functional and regulatory effect of genetic variants identified from the current GWAS in a range of tissues was measured systematically using GARFIELD, (Iotchkova et al., 2016) as a novel enrichment approach that leverages GWAS findings with regulatory functional annotations. GARFIELD quantifies fold enrichment (FE) for that annotation, while taking into account LD, MAF and local gene densities. GWAS SNPs are reduced to an independent set by pruning to retain only the most significant variants (LD r2 > 0.1 and within 1-Mb window), which are annotated if they, or their LD proxies (LD r2 > 0.8), have regulatory information overlapping with functional features (mainly derived from the Encyclopaedia of DNA Elements [ENCODE] and Roadmap Epigenomics data). FE statistics were calculated by including genetic variants at seven genome-wide significance thresholds (10−1 to 10−7), among which the significances of FE at four GWAS p value thresholds 10−4 to 10−7 were analysed by permutation testing. Multiple testing correction was further performed based on the effective number of annotations.
Results
Following quality control and genotype imputation, data were available on 6,473,404 markers genome wide in 684 individuals. We performed logistic regression to test genetic associations with schizophrenia among 429 cases and 255 controls, adjusting for gender and population structure using the first two ancestry-informative eigenvectors generated from PCA. The strongest association with schizophrenia in GWAS was observed for the SNP rs10252923 (p = 2.01 × 10−6, odds ratio [OR] = 1.82, 95% confidence interval [CI] = [1.42, 2.33]) located at 7q21.13, within the downstream region of frizzled class receptor 1 (FZD1) and 65 kb away from cyclin-dependent kinase 14 (CDK14) (Figure 1) (Table 1). Regional plot by LocusZoom (Pruim et al., 2010) was performed for this region (Figure 2). Conditional analysis was performed in this locus, and no independent secondary associations with p < 1.00 × 10−5 were found.
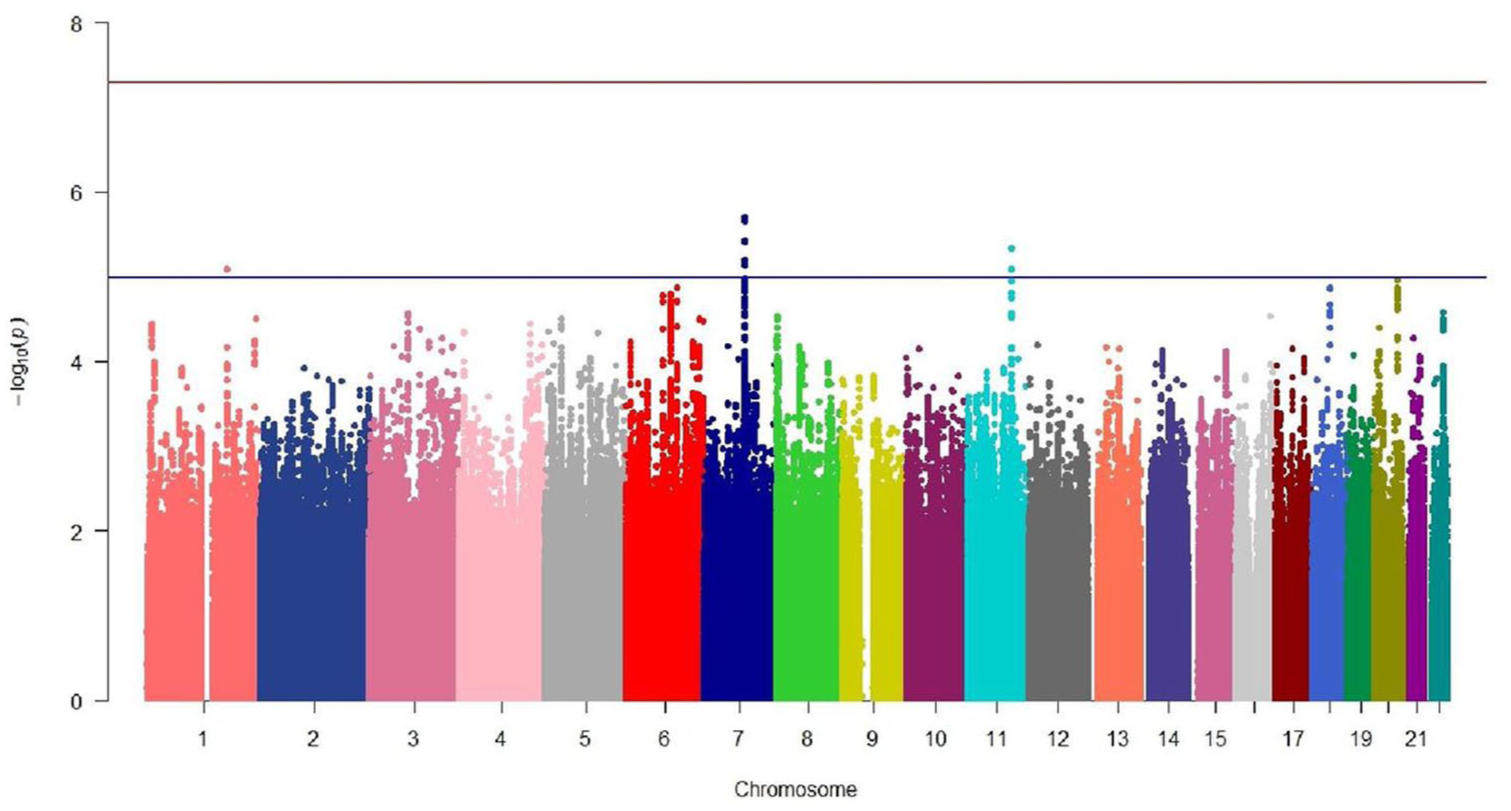
A Manhattan plot showing schizophrenia genome-wide associations (429 cases and 255 controls). The x-axis is chromosomal position, and the y-axis is the significance (−log10 P; two tailed) of association derived by logistic regression. The red line denotes the genome-wide significance threshold (5 × 10−8), and the blue line shows the genome-wide suggestive significance level (1 × 10−5).
Top five strongest SNP GWAS signals.
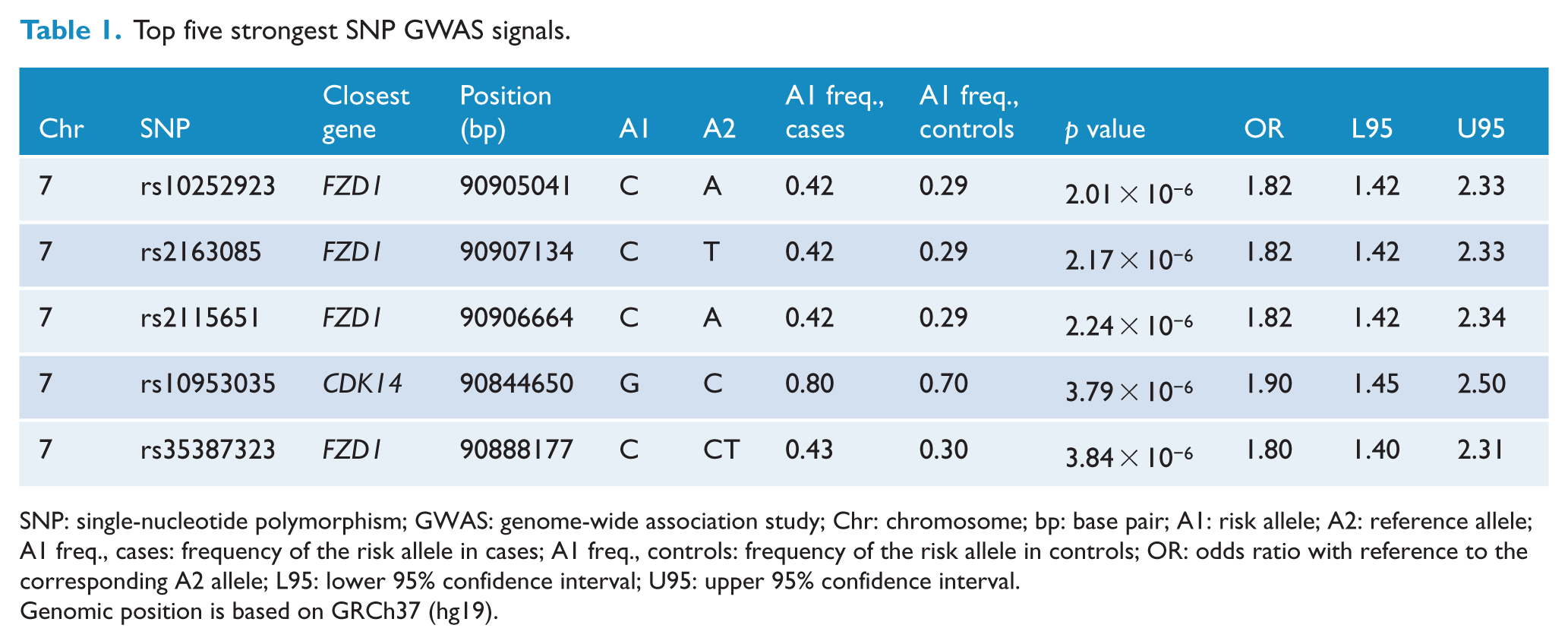
SNP: single-nucleotide polymorphism; GWAS: genome-wide association study; Chr: chromosome; bp: base pair; A1: risk allele; A2: reference allele; A1 freq., cases: frequency of the risk allele in cases; A1 freq., controls: frequency of the risk allele in controls; OR: odds ratio with reference to the corresponding A2 allele; L95: lower 95% confidence interval; U95: upper 95% confidence interval.
Genomic position is based on GRCh37 (hg19).
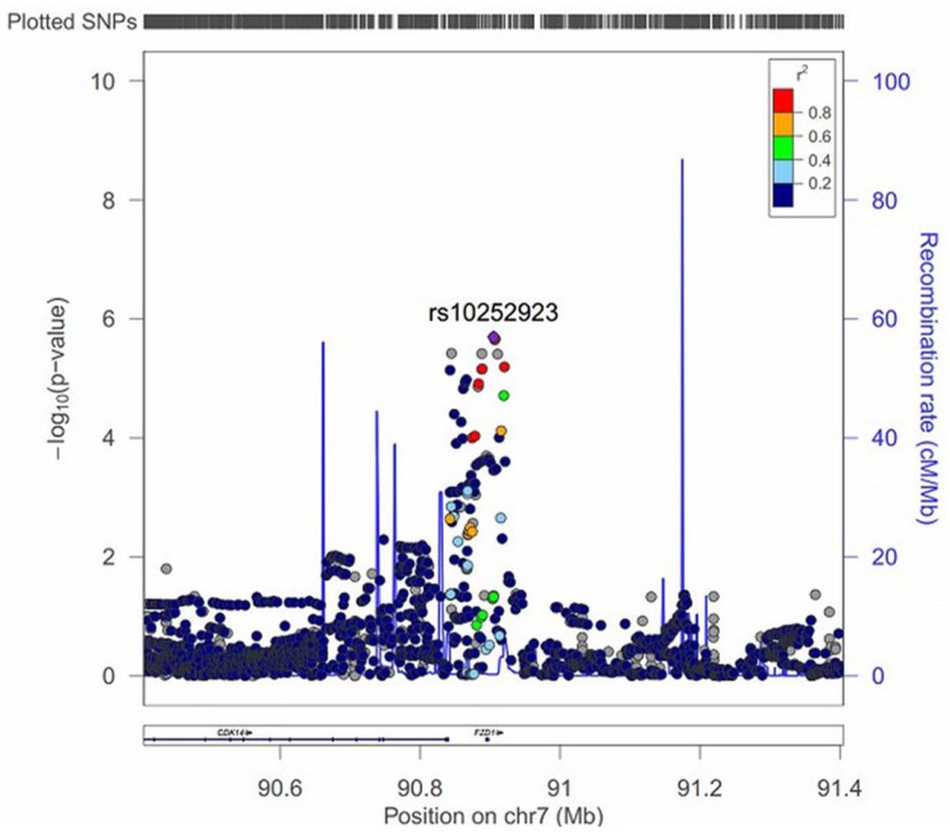
Regional association plot for associations with schizophrenia at locus 7q21.13 (FZD1, frizzled class receptor 1, and CDK14, cyclin-dependent kinase 14). The x-axis represents chromosomal position, and the y-axis indicates the significance (−log10 P; two tailed) of association derived by logistic regression. Diamond denotes the lead variant (with the most significant p value), and circles show genotyped variants, while crosses shows imputed variants. The plot includes recombination rates (light blue peaks) and the locations of genes (dark blue horizontal lines in the bottom).
Gene-based analysis considers the impact of SNPs within a single gene, which individually may not attain statistical significance in GWAS. Applying MAGMA to our genome-wide association data identified FZD1 (p = 2.78 × 10−6) as meeting the significance threshold (Table 2). This stringent Bonferroni threshold is likely to be overly conservative, indicating the data provided good evidence for an association between FZD1 and schizophrenia. The top signal in MAGENTA analysis was ionotropic glutamate receptor pathway, with a nominal p value of 1.30 × 10−3, but the adjusted p value after correction for multiple testing (FDR = 8.66 × 10−2) did not meet the significance threshold (Supplementary Table 1).
MAGMA gene-based association of the top five results.
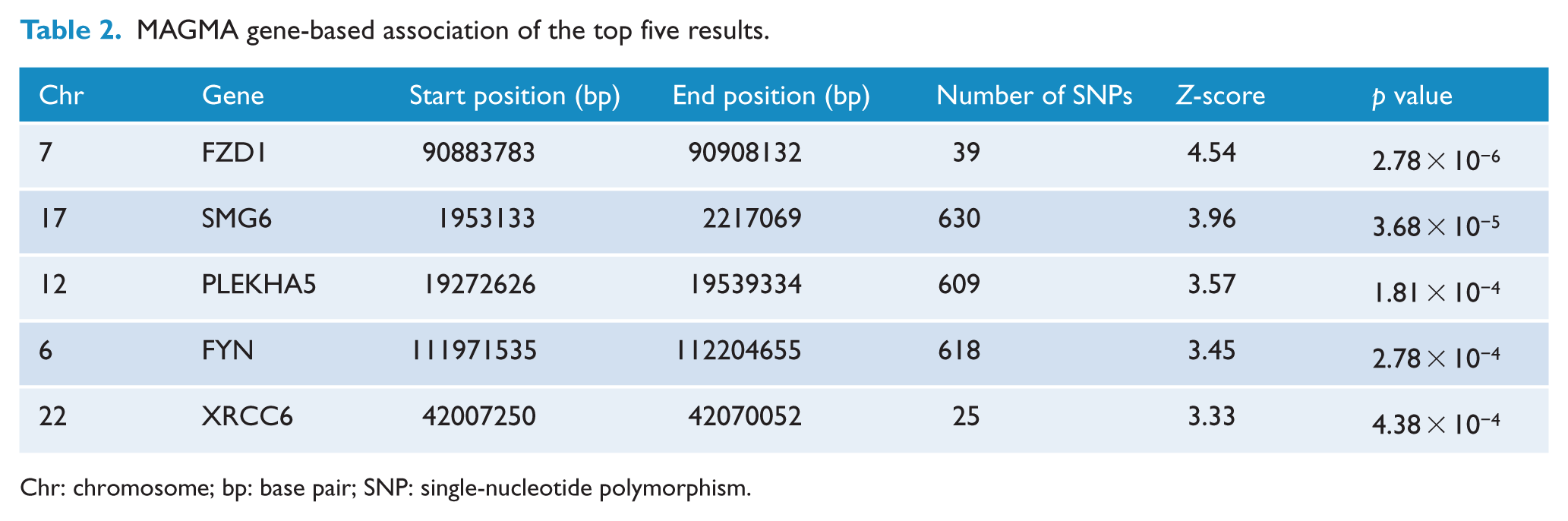
Chr: chromosome; bp: base pair; SNP: single-nucleotide polymorphism.
To elucidate potential functional consequences of the genetic marker–mediated change in gene expression, we conducted eQTL analyses using the GTEx portal. This identified rs10252923 to have nominal cis-eQTL effects on nearby genes, FZD1 and CDK14, in different tissues (Supplementary Table 2, Supplementary Figure 3). In particular, it had nominal association with CDK14 expression in the nucleus accumbens (p = 0.047).
The enrichment analysis of regulatory and functional features was performed using GARFIELD and interrogated sets of independent SNPs co-localized and overrepresented within functional regions in different tissues. Approximately threefold enrichment (significant FE) for DNase I hypersensitive site (DHS) hotspots in foetal brain tissue was observed, when genetic variants with p values < 10−4 were included. The enrichment levels of GWAS SNPs with p values < 10−4 increased greatly over those of SNPs with p values < 10−3 (Supplementary Figure 4).
Discussion
This study comprehensively analysed common variant risk for schizophrenia in the ASRB cohort. Given the relatively small sample size and the genetic complexity of the disorder, identification of variants of genome-wide significance was not expected. While this was confirmed, our functional characterization of the variant distribution provides new biological insight into the genomic risk of schizophrenia with the identification of functional variants and genes associated with the disorder.
At the variant level, the top GWAS signals were located at 7q21.13 in two genes, FZD1 and CDK14. In a post-GWAS analysis, FZD1, at the gene level, was also enriched for its involvement in the susceptibly to schizophrenia via gene-based analysis (p = 2.78 × 10−6). Gene-based analysis relies on converging evidence from multiple genetic markers in the gene, with the assumption that disease-associated risk alleles will converge on genes with biological significance. It can identify biologically oriented association signals that are not necessarily revealed by conventional variant-level analysis. This is, especially, the case for complex diseases like schizophrenia where susceptibility is distributed across multiple genetic variants with small to moderate effect sizes.
Neither FZD1 nor CDK14 has been implicated in schizophrenia previously, and the closest gene reported to be significantly associated with the disease in a genome-wide analysis was glutamate metabotropic receptor 3 (GRM3) located at 7q21.12 (Goes et al., 2015). FZD1 is involved in the Wnt signalling pathway, which is activated by the interaction of Wnt ligands with members of the frizzled family of seven-transmembrane domain cell surface receptors (Logan and Nusse, 2004). The Wnt signalling network is of great complexity, contains various molecular components and is subject to a series of regulatory steps and cross-talks. The major signalling branches include the canonical Wnt/β-catenin pathway and the non-canonical Wnt/planar cell polarity and Wnt/calcium pathways (Komiya and Habas, 2008). Wnt signalling pathway is essential for neurological development and the maintenance of the nervous system (Moon et al., 2004; Okerlund and Cheyette, 2011; Panaccione et al., 2013; Patapoutian and Reichardt, 2000) and has been implicated in several neuropsychiatric diseases, including schizophrenia and bipolar disorder (Miyaoka et al., 1999; Okerlund and Cheyette, 2011; Panaccione et al., 2013). FZD1 deletion mutants showed markedly reduced capacity to enhance Wnt signal transduction (Gazit et al., 1999). FZD1 can interact with several Wnt proteins (1, -2, -3, -3a, -5a and -7b) (Dijksterhuis et al., 2014), and an increase of Wnt-1 immunoreactivity was found in postmortem hippocampus in schizophrenia compared to non-psychiatric controls (Miyaoka et al., 1999). Furthermore, knocking down of FZD1 expression decreased the protective effect of Wnt-3a on hippocampal neurons against the toxicity of amyloid-β-peptide (Chacon et al., 2008), and activation of Wnt-5a enhances GABA efficacy (Cuitino et al., 2010). Our finding provides genetic support for FZD1 involvement in schizophrenia.
CDK14 is in close proximity to FZD1 and is also involved in the Wnt pathway (Logan and Nusse, 2004). CDK14 regulates Wnt/β-catenin signalling through mediation of cell cycle–dependent lipoprotein receptor–related protein 6 (LRP6) phosphorylation (Davidson et al., 2009; Davidson and Niehrs, 2010). As polymorphisms at the DNA level may lead to changes in intermediate quantifiable phenotypes (e.g. RNA and protein expression level), we used the GTEx portal to explore eQTL and the functional mechanisms of the top SNPs identified in our study. Even though rs10252923 did not surpass genome-wide significance, this SNP was the strongest signal detected at the variant level and showed nominal association with the expression levels of two neighbouring genes, FZD1 and CDK14, in tissues including the nucleus accumbens of the brain.
Further functional integration of common variation across the genome with respect to chromatin annotation was performed using GARFIELD. This revealed significant enrichment of SNPs in DHS associated with foetal brain tissues (Sheffield and Furey, 2012). This supports a role for variation of regulatory motifs in chromatin involved in foetal brain development in schizophrenia. The significance of enrichment was observed for SNPs with GWAS significance p < 10−4, further indicating the genetic complexity of the disorder. The hypothesis that genomic variation is associated with altered neurodevelopment is supported by several observations and is consistent with early adverse environmental exposures, such as maternal infection during pregnancy, which also negatively influences brain development and confers an increased risk of schizophrenia (Brown et al., 2004; Ellman et al., 2010; Meyer et al., 2009; Urakubo et al., 2001).
Conclusion
We conducted a GWAS of schizophrenia in the ASRB cohort. While no individual variants reached genome-wide significance, integration of independent association signal at the gene level by MAGMA implicated FZD1, which encodes a receptor protein involved in the Wnt signalling pathway. SNPs were also enriched in open chromatin regions associated with foetal brain development, suggesting these variants have regulatory function in neurodevelopment.
Supplemental Material
Supplementary_Figure_1A – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_Figure_1A for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_Figure_1B – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_Figure_1B for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_Figure_2 – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_Figure_2 for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_Figure_3 – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_Figure_3 for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_Figure_4 – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_Figure_4 for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_files_of_GWAS_of_schizophrenia_684indiv_EUR_3covariates_v3-revised_v1 – Supplemental material for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort
Supplemental material, Supplementary_files_of_GWAS_of_schizophrenia_684indiv_EUR_3covariates_v3-revised_v1 for Wnt receptor gene FZD1 was associated with schizophrenia in genome-wide SNP analysis of the Australian Schizophrenia Research Bank cohort by Xiaoman Liu, Siew-Kee Low, Joshua R Atkins, Jing Qin Wu, William R Reay, Heath M Cairns, Melissa J Green, Ulrich Schall, Assen Jablensky, Bryan Mowry, Patricia T Michie, Stan V Catts, Frans Henskens, Christos Pantelis, Carmel Loughland, Alan V Boddy, Paul A Tooney, Rodney J Scott, Vaughan J Carr and Murray J Cairns in Australian & New Zealand Journal of Psychiatry
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Data and samples were collected by the Australian Schizophrenia Research Bank (ASRB), supported by the Australian NHMRC, the Pratt Foundation, Ramsay Health Care and the Viertel Charitable Foundation. The ASRB was also supported by the Schizophrenia Research Institute (Australia), utilizing infrastructure funding from the NSW Health and the Macquarie Group Foundation. DNA analysis was supported by the Neurobehavioral Genetics Unit, utilizing funding from NSW Health. J.R.A. was supported by the University of Newcastle RHD and an Emlyn and Jennie Thomas Postgraduate Medical Research Scholarship. W.R.R. is supported by an Australian Postgraduate Award. C.L. was supported by National Health and Medical Research Council (NHMRC) Project Grants (1067137). M.J.C. was supported by an NHMRC Senior Research Fellowship (1121474). C.P. was supported by a NHMRC Senior Principal Research Fellowship (628386 & 1105825).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.